CD23 is the low affinity receptor for IgE. When it appears on eosinophils and macrophages it helps these cells to kill and eat structures that are coated with IgE antibody, but this isn't what it does on CLL cells. Some have suggested that it is a growth factor for B cells, but this is disputed. Others have suggesetd it binds IgE immune complexes and this increases the efficiency of B cell antigen presentation and processing. I can't see why this is important for CLL cells. We also know that it is associated with the fyn tyrosine kinase.
It is present on CLL cells, but not usually on other types of B cell lymphoma cells. It is also present in the serum of CLL patients. In continental Europe, particularly Italy, it has been used as a prognostic factor for CLL. It has not traveled. I think the main problem with it is that it is a surrogate marker for both the amount of CLL cells as well as their growth rate, and therefore not particularly useful for predicting the future.
Saturday, December 31, 2005
The perfect forward defensive stroke
Body memory is a strange thing. When asked to produce their signature using a broom in snow, volunteers produced a scaled up version of the real thing, even though a completely different set of muscles was involved. When I close my eyes I can feel myself in the position of the forward defensive stroke even though I haven't picked up a cricket bat in ten years.
The forward defensive stroke was the only stroke I mastered. The sports reports would enthuse about Wally Hammond's cover drive or Dennis Compton's sweep, but I was besotted by the forward defensive stroke. I raved about Peter Richardson's 117 at Port Elizabeth. Occupying the crease seemed to me to be the greatest ambition.
The balance has to be right. Up on the balls of your feet, never back on your heels; you take a good stride forward. It takes courage against the quicks. Your natural instinct is to back away; a ball in the face the reward for foolhardiness. The stride should stretch the hamstrings of the back leg. Little back lift, just enough to bring the bat down straight, you lead with your left elbow pointed. Your weight goes forward, your head over the ball, and still. You have watched the ball leave the bowler's hand. You must not blink. Your shin determines the line of the stroke. Bat and pad together. "Bat and pad together!" The instructor's cry. There is the danger. Do not open 'the gate' for the one that nips back. Know where your off stump is. This far forward you won't be LBW. Don't worry about the ball that rises and hits you; it can't get you out and bruises fade. Get close to the pitch of the ball. The closer you are, the less it can do off the seam. Watch for the one swings away. Do not follow it. DO NOT FOLLOW IT! It is missing off stump. He is trying to catch the edge. And then the satisfying 'chock' of leather on willow. Keep soft hands and the bat angled down. The ball is smothered and the bowler frustrated. He will call something rude at you. You don't listen. You look up at him and grin to rub it in.
The forward defensive stroke was the only stroke I mastered. The sports reports would enthuse about Wally Hammond's cover drive or Dennis Compton's sweep, but I was besotted by the forward defensive stroke. I raved about Peter Richardson's 117 at Port Elizabeth. Occupying the crease seemed to me to be the greatest ambition.
The balance has to be right. Up on the balls of your feet, never back on your heels; you take a good stride forward. It takes courage against the quicks. Your natural instinct is to back away; a ball in the face the reward for foolhardiness. The stride should stretch the hamstrings of the back leg. Little back lift, just enough to bring the bat down straight, you lead with your left elbow pointed. Your weight goes forward, your head over the ball, and still. You have watched the ball leave the bowler's hand. You must not blink. Your shin determines the line of the stroke. Bat and pad together. "Bat and pad together!" The instructor's cry. There is the danger. Do not open 'the gate' for the one that nips back. Know where your off stump is. This far forward you won't be LBW. Don't worry about the ball that rises and hits you; it can't get you out and bruises fade. Get close to the pitch of the ball. The closer you are, the less it can do off the seam. Watch for the one swings away. Do not follow it. DO NOT FOLLOW IT! It is missing off stump. He is trying to catch the edge. And then the satisfying 'chock' of leather on willow. Keep soft hands and the bat angled down. The ball is smothered and the bowler frustrated. He will call something rude at you. You don't listen. You look up at him and grin to rub it in.
Friday, December 30, 2005
Revlimid
Revlimid is an analogue of thalidomide that has been developed as part of a research programme to separate the evil effects of thalidomide from the beneficial effects. Are there any beneficial effects?
Well, in multiple myeloma, in patients who had failed even the so called "total therapy" use in Little Rock, there was a 37% response rate when treated with thalidomide. It has also been used in some other cancers, producing remissions where there was previously no hope. Thalidomide has also been useful in some forms of Leprosy and in some types of Graft-versus-Host disease. Even without the birth defects, thalidomide is still an unpleasant drug. It cause a peripheral neuropathy, severe constipation and sleepiness. In myeloma it increases the rate at which venous thrombosis and pulmonary embolus occur. But, of course, the major reason for being wary of it are the birth defects that it caused, babies born without arms or legs (known as phocomelia). This was perhaps the greatest drug scandal ever.
Does Revlimid have the same side effects?
Not surprisingly it has never been given to pregnant women. Thalidomide was never tested in pregnant animals before it was used, but after the birth defects were found, it turned out that it was possible to reproduce them in pregnant animals. The pregnant animal test is now a must for all drug testing. In pregnant animal tests that produce the birth defects with thalidomide, it has not been found that Revlimid is free of this side effect. Developmental toxicology studies were conducted in rats and rabbits to examine possible teratogenic effects of lenalidomide. The rabbit study contained a thalidomide arm as a positive control, as the New Zealand White rabbit is known to be sensitive to thalidomide’s teratogenic effects. Teratogenic effects were not seen in either study with lenalidomide. However, the rat is
not sensitive to thalidomide and thus is not considered a useful model for evaluating
thalidomide-like effects. The highest dose used in the pivotal rabbit teratogenicity study did not meet the level of being sufficiently maternally toxic, a standard endpoint in teratogenicity studies to assess appropriate dosing. Maternal toxicity was observed in rabbits at higher doses in the dose-range finding study. It has been given to pregnant rats at 600 times the dose to be used in humans and no fetal abnormalities were found. In mice, doses of 100 times that to be used in humans killed the embryos. But there was no phocomelia.
Thalidomide is a well-known teratogen, but the mechanism of teratogenicity is not established. It is not known whether thalidomide itself, degradation product(s), or both are responsible for teratogenicity. Thalidomide derived products have been identified in animals and humans; lenalidomide derived products have been identified in animals but not searched for in humans. It is likely that both compounds share similar metabolic or degradative pathways. Modeling suggests that the intermediates and final products would be structurally similar, but chemically unique, for each drug.
Does that mean that it is safe to use in pregnant women? By no means. I doubt that any thalidomide analogue will ever be licensed to be used in pregnancy. Indeed the conditions of the license are that special precautions must be taken to avoid it being given to pregnant women inadvertently.
What about the other side effects?
It does not cause sleepiness, neuropathy or constipation. But clinical trials have uncovered other problems. It causes diarrhea, though this can be overcome by appropriate treatment. Most important, in MDS it causes bone marrow suppression. This does not seem to be a major problem in multiple myeloma, where there is a lot of experience of using a larger dose, but in MDS the bone marrow cells are damaged to start with, whereas in myeloma the marrow is occupied by the myeloma, but the normal bone marrow cells are not themselves damaged.
The license is for MDS with deletion 5q. The clinical trial involved 148 MDS patients who all had a part of their long arm of chromosome 5 missing. Now there is confusion about this condition. Not everyone with MDS and a missing 5q has the 5q minus syndrome. 5q minus syndrome is a fairy benign form of MDS occurring mainly in older females, who have macrocytosis and a normal or high platelet count, and in the marrow, mononuclear megakaryocytes. The patients in the trial included some of these, but many who just had low or intermediate grade MDS with a missing 5q. 34.5% were males, 25% had other chromosomal abnormalities as well, 48% had RARS, RAEB, or CMML, all had a transfusion requirement. So in evaluating the results it is wrong to think that this was the rather benign 5q minus syndrome that often does require therapy.
In all clinical trials of effective chemotherapy in MDS marrow suppression is expected. Although Revlimid has many other effects, in MDS it seems to act primarily as a cytotoxic drug, rather than a supportive care drug like erythropoietin (EPO). EPO will produce relief from the need for transfusion in 30-50% of low and intermediate grade I MDS, but it will not get rid of the abnormal clone. On the other hand in this trial Revlimid relieved the need for transfusion in 67% and got rid of the abnormal clone in a substantial proportion of patients. Cytotoxic drugs produce severe cytopenias lasting in excess of 3 weeks in MDS, and in MDS Revlimid caused severe neutropenia or thrombocytopenia in 53.4% and 50% respectively. Febrile neutropenia (getting a high temperature while the patient has low white cells) occurred in 5.4%, which is very low for a cytotoxic drug. 11.5% got pneumonia. There were 11 deaths in the trial. 2 of these were attributed to the Revlimid. Now you may argue that these deaths have not been independently evaluated and that the other 9 that were attributed to the disease might have been contributed to by the drug. All I can say that the FDA had more information than we have on that and they have licensed it for this use. The FDA is very touchy on thalidomide analogues, and it seems to me that they would be unlikely to approve one that relied on camouflaged data.
So how does this relate to CLL?
Revlimid has not been licensed for use in CLL and neither has thalidomide. However in small phase II trials both have been shown to have some activity in CLL. Do not be persuaded to take either of these drugs unlicensed, except in the context of a clinical trial. Clinical trials have to be assessed by ethical committees. They will regard any trial as unethical if the risk to the patient is greater for taking the drug rather than avoiding it. Therefore there will be strict entry criteria for these trials, and patients entered into the trials will be closely monitored. Ideally these trials should be randomized phase III trials. If we had these for MDS we would know for certain whether the 11 deaths were caused by the disease or the treatment.
Well, in multiple myeloma, in patients who had failed even the so called "total therapy" use in Little Rock, there was a 37% response rate when treated with thalidomide. It has also been used in some other cancers, producing remissions where there was previously no hope. Thalidomide has also been useful in some forms of Leprosy and in some types of Graft-versus-Host disease. Even without the birth defects, thalidomide is still an unpleasant drug. It cause a peripheral neuropathy, severe constipation and sleepiness. In myeloma it increases the rate at which venous thrombosis and pulmonary embolus occur. But, of course, the major reason for being wary of it are the birth defects that it caused, babies born without arms or legs (known as phocomelia). This was perhaps the greatest drug scandal ever.
Does Revlimid have the same side effects?
Not surprisingly it has never been given to pregnant women. Thalidomide was never tested in pregnant animals before it was used, but after the birth defects were found, it turned out that it was possible to reproduce them in pregnant animals. The pregnant animal test is now a must for all drug testing. In pregnant animal tests that produce the birth defects with thalidomide, it has not been found that Revlimid is free of this side effect. Developmental toxicology studies were conducted in rats and rabbits to examine possible teratogenic effects of lenalidomide. The rabbit study contained a thalidomide arm as a positive control, as the New Zealand White rabbit is known to be sensitive to thalidomide’s teratogenic effects. Teratogenic effects were not seen in either study with lenalidomide. However, the rat is
not sensitive to thalidomide and thus is not considered a useful model for evaluating
thalidomide-like effects. The highest dose used in the pivotal rabbit teratogenicity study did not meet the level of being sufficiently maternally toxic, a standard endpoint in teratogenicity studies to assess appropriate dosing. Maternal toxicity was observed in rabbits at higher doses in the dose-range finding study. It has been given to pregnant rats at 600 times the dose to be used in humans and no fetal abnormalities were found. In mice, doses of 100 times that to be used in humans killed the embryos. But there was no phocomelia.
Thalidomide is a well-known teratogen, but the mechanism of teratogenicity is not established. It is not known whether thalidomide itself, degradation product(s), or both are responsible for teratogenicity. Thalidomide derived products have been identified in animals and humans; lenalidomide derived products have been identified in animals but not searched for in humans. It is likely that both compounds share similar metabolic or degradative pathways. Modeling suggests that the intermediates and final products would be structurally similar, but chemically unique, for each drug.
Does that mean that it is safe to use in pregnant women? By no means. I doubt that any thalidomide analogue will ever be licensed to be used in pregnancy. Indeed the conditions of the license are that special precautions must be taken to avoid it being given to pregnant women inadvertently.
What about the other side effects?
It does not cause sleepiness, neuropathy or constipation. But clinical trials have uncovered other problems. It causes diarrhea, though this can be overcome by appropriate treatment. Most important, in MDS it causes bone marrow suppression. This does not seem to be a major problem in multiple myeloma, where there is a lot of experience of using a larger dose, but in MDS the bone marrow cells are damaged to start with, whereas in myeloma the marrow is occupied by the myeloma, but the normal bone marrow cells are not themselves damaged.
The license is for MDS with deletion 5q. The clinical trial involved 148 MDS patients who all had a part of their long arm of chromosome 5 missing. Now there is confusion about this condition. Not everyone with MDS and a missing 5q has the 5q minus syndrome. 5q minus syndrome is a fairy benign form of MDS occurring mainly in older females, who have macrocytosis and a normal or high platelet count, and in the marrow, mononuclear megakaryocytes. The patients in the trial included some of these, but many who just had low or intermediate grade MDS with a missing 5q. 34.5% were males, 25% had other chromosomal abnormalities as well, 48% had RARS, RAEB, or CMML, all had a transfusion requirement. So in evaluating the results it is wrong to think that this was the rather benign 5q minus syndrome that often does require therapy.
In all clinical trials of effective chemotherapy in MDS marrow suppression is expected. Although Revlimid has many other effects, in MDS it seems to act primarily as a cytotoxic drug, rather than a supportive care drug like erythropoietin (EPO). EPO will produce relief from the need for transfusion in 30-50% of low and intermediate grade I MDS, but it will not get rid of the abnormal clone. On the other hand in this trial Revlimid relieved the need for transfusion in 67% and got rid of the abnormal clone in a substantial proportion of patients. Cytotoxic drugs produce severe cytopenias lasting in excess of 3 weeks in MDS, and in MDS Revlimid caused severe neutropenia or thrombocytopenia in 53.4% and 50% respectively. Febrile neutropenia (getting a high temperature while the patient has low white cells) occurred in 5.4%, which is very low for a cytotoxic drug. 11.5% got pneumonia. There were 11 deaths in the trial. 2 of these were attributed to the Revlimid. Now you may argue that these deaths have not been independently evaluated and that the other 9 that were attributed to the disease might have been contributed to by the drug. All I can say that the FDA had more information than we have on that and they have licensed it for this use. The FDA is very touchy on thalidomide analogues, and it seems to me that they would be unlikely to approve one that relied on camouflaged data.
So how does this relate to CLL?
Revlimid has not been licensed for use in CLL and neither has thalidomide. However in small phase II trials both have been shown to have some activity in CLL. Do not be persuaded to take either of these drugs unlicensed, except in the context of a clinical trial. Clinical trials have to be assessed by ethical committees. They will regard any trial as unethical if the risk to the patient is greater for taking the drug rather than avoiding it. Therefore there will be strict entry criteria for these trials, and patients entered into the trials will be closely monitored. Ideally these trials should be randomized phase III trials. If we had these for MDS we would know for certain whether the 11 deaths were caused by the disease or the treatment.
Wednesday, December 28, 2005
Beta-2 microglobulin
Beta-2 microglobulin is used by some centers as a prognostic factor and is particularly favored by the MD Anderson Cancer Center in Houston.
Bete-2 microglobulin is the small subunit of the MHC class I molecule. The MHC class I molecule is the same as the HL-A antigen that is present on the surface of almost all nucleated cells in the body. These HL-A antigens are the expression of self for every cell and it is because they are recognised as such that they and the peptides they carry are targeted by a foreign immune system. The beta 2-microglobulin is generally required for the transport of class I heavy chains from the endoplasmic reticulum (endoplasmic reticulum is the factory floor of the cell) to the cell surface.
Beta 2-microglobulin is present in small amounts in serum, and urine of normal people,the normal level varies from lab to lab, but up to 2.7 mg/dL is a figure given by Google. There is a lot of beta-2 M on lymphocytes. Therefore, anything that makes lymphocytes multiply can give a raised level. Virus infections, particularly infectious mononucleosus and CMV infections can do this.
The level of beta-2 M is one of the best prognostic factors for multiple myeloma but this exposes one of its problems. Renal damage is common in myeloma, but beta-2 M is excreted through the kidneys so there is a risk of a falsely high value. In lymphomas beta-2 M has been used as a prognostic factor as it has in CLL, but it measures two separate things: a] the amount of lymphoma or CLL present, and b] the rate of turnover of lymphoma or CLL cells. In early stage CLL it has never been tested as a means of predicting who in the future will need treatment.
There is no reference to turn to for how well beta-2 M performs as a prognostic factor in CLL. Although in terms of ease of use it has advantages few centers have adopted it.
Bete-2 microglobulin is the small subunit of the MHC class I molecule. The MHC class I molecule is the same as the HL-A antigen that is present on the surface of almost all nucleated cells in the body. These HL-A antigens are the expression of self for every cell and it is because they are recognised as such that they and the peptides they carry are targeted by a foreign immune system. The beta 2-microglobulin is generally required for the transport of class I heavy chains from the endoplasmic reticulum (endoplasmic reticulum is the factory floor of the cell) to the cell surface.
Beta 2-microglobulin is present in small amounts in serum, and urine of normal people,the normal level varies from lab to lab, but up to 2.7 mg/dL is a figure given by Google. There is a lot of beta-2 M on lymphocytes. Therefore, anything that makes lymphocytes multiply can give a raised level. Virus infections, particularly infectious mononucleosus and CMV infections can do this.
The level of beta-2 M is one of the best prognostic factors for multiple myeloma but this exposes one of its problems. Renal damage is common in myeloma, but beta-2 M is excreted through the kidneys so there is a risk of a falsely high value. In lymphomas beta-2 M has been used as a prognostic factor as it has in CLL, but it measures two separate things: a] the amount of lymphoma or CLL present, and b] the rate of turnover of lymphoma or CLL cells. In early stage CLL it has never been tested as a means of predicting who in the future will need treatment.
There is no reference to turn to for how well beta-2 M performs as a prognostic factor in CLL. Although in terms of ease of use it has advantages few centers have adopted it.
Tuesday, December 27, 2005
Other prognostic factors - doubling time
Doubling time.
This is a simple measurement of how rapidly the CLL is growing. A lymphocyte count doubling every six months is an indication for treatment according to the NCI, though some trials have adopted a less stringent criterion - doubling every 12 months - as an indication for treatment (for example the British CLL4 trial).
How accurately does the doubling time reflect the true growth in the size of the leukemic clone? While the count is very low it does not really do so at all. Most of the growth takes place within the bone marrow, and the spillover element in the blood is variable. As the count increases it is more accurate, but it has to be remembered that within the lymphocyte count there is a variable number of T cells and NK cells. Only when the count is fairly high, say over 30k is it safe to neglect this.
It is important to exclude the other variables. Strange to say it should not be assumed that different laboratories will give the same answer. Even very famous epartment can get things wrong. The truth is that the famous head of department is usually traveling from meeting to meeting giving presentations or sitting on committees deciding on grants. You can certainly rely on his research laboratory, because the very best people will be competing to work for him (or her), but the routine laboratory in his hospital is unlikely to be under his day to day control. If the laboratory is in a large city, it may be difficult to find laboratory technicians to run the machines - it is not a well paid job, and there are plenty of jobs in the city that are well paid.
In one of the tasks I undertook when I was employed by the National Health Service I had to go around inspecting other people's laboratories. I usually got landed with the London Teaching Hospitals. Surprisingly, they were often worse than ordinary laboratories at ordinary hospitals. In one blood transfusion laboratory where the person who had written a well known text book worked, entirely the wrong concentration of suspended red cells was being used. In another, where a well known expert in laborartory technology was in charge, the condensors on the laboratory microscopes couldn't be adjusted because of all the crud on them. So it was no surprise to me when I was sent a sequence of lymphocyte counts which showed an apparent doubling thast was entirely due to a rogue result from a famous laboratory.
One technical problem is that the machines are callibrated to give an accurate count in the normal - high range , but when the count is very high it becomes progressively inaccurate. The sample then has to be diluted to get the count back into the accurate range. This dilution introduces a potential source of error.
It also has to be remembered that the lymphocyte count goes up an down according to other things than progression of the CLL. Following an infection the lymphocyte count rises as the immune response kicks in. this effect is emphasized by the crowding out of normal lymphocytes from the lymph nodes by the infiltrating CLL cells. Vaccination also induces an immune response, so it can also raise the lymphocyte count transiently.
For all these reasons it is important not to accept a single observation as evidence of progression. Unless the count is rising rapidly, I would expect to follow a patients for 3-6 months with several blood tests before being convinced of a rapid lymphocyte doubling time.
This is a simple measurement of how rapidly the CLL is growing. A lymphocyte count doubling every six months is an indication for treatment according to the NCI, though some trials have adopted a less stringent criterion - doubling every 12 months - as an indication for treatment (for example the British CLL4 trial).
How accurately does the doubling time reflect the true growth in the size of the leukemic clone? While the count is very low it does not really do so at all. Most of the growth takes place within the bone marrow, and the spillover element in the blood is variable. As the count increases it is more accurate, but it has to be remembered that within the lymphocyte count there is a variable number of T cells and NK cells. Only when the count is fairly high, say over 30k is it safe to neglect this.
It is important to exclude the other variables. Strange to say it should not be assumed that different laboratories will give the same answer. Even very famous epartment can get things wrong. The truth is that the famous head of department is usually traveling from meeting to meeting giving presentations or sitting on committees deciding on grants. You can certainly rely on his research laboratory, because the very best people will be competing to work for him (or her), but the routine laboratory in his hospital is unlikely to be under his day to day control. If the laboratory is in a large city, it may be difficult to find laboratory technicians to run the machines - it is not a well paid job, and there are plenty of jobs in the city that are well paid.
In one of the tasks I undertook when I was employed by the National Health Service I had to go around inspecting other people's laboratories. I usually got landed with the London Teaching Hospitals. Surprisingly, they were often worse than ordinary laboratories at ordinary hospitals. In one blood transfusion laboratory where the person who had written a well known text book worked, entirely the wrong concentration of suspended red cells was being used. In another, where a well known expert in laborartory technology was in charge, the condensors on the laboratory microscopes couldn't be adjusted because of all the crud on them. So it was no surprise to me when I was sent a sequence of lymphocyte counts which showed an apparent doubling thast was entirely due to a rogue result from a famous laboratory.
One technical problem is that the machines are callibrated to give an accurate count in the normal - high range , but when the count is very high it becomes progressively inaccurate. The sample then has to be diluted to get the count back into the accurate range. This dilution introduces a potential source of error.
It also has to be remembered that the lymphocyte count goes up an down according to other things than progression of the CLL. Following an infection the lymphocyte count rises as the immune response kicks in. this effect is emphasized by the crowding out of normal lymphocytes from the lymph nodes by the infiltrating CLL cells. Vaccination also induces an immune response, so it can also raise the lymphocyte count transiently.
For all these reasons it is important not to accept a single observation as evidence of progression. Unless the count is rising rapidly, I would expect to follow a patients for 3-6 months with several blood tests before being convinced of a rapid lymphocyte doubling time.
Monday, December 26, 2005
Christmas Day
They say that Christmas is for the children. This has been the first Christmas day without little children in my life. The youngest was 25. All through my childhood I had siblings (except in 1943, but I spent that in my grandmother's house and I expect that there were cousins about. I was too young to remember). In 1967 when we were first married my parent's in-law spent Christmas with us and brought my wife's sister who was just a little girl. In 1994 the youngest child present was 14, and ever since we have had grandchildren with us. We will see the grandchildren on Wednesday, but this Christmas was child-free. It was also the most stress-free ever. Nobody wouldn't eat her dinner, or broke his toy, or cried because the battery was lacking.
Presents were unexpected and funny. I had an enormous kipper tie that is held on by an elastic band, a mounted football shirt of Aldershot Town, the club for which I was a mascot during the 1950-51 season and a full set of the Sharpe DVDs. I gave my wife specialty herbal teas, a really expensive jar of lemon curd, and a number of biographies including one of Philipp Bliss, the author of "Man of Sorrows".
We played 3 games of Scrabble, which everybody won except me. We opened a bottle of champagne that had been given to my daughter, the doctor, by a patient, and in between eating (the food was excellent) I worked on a new carol. I have never really liked the words of O Holy Night. The tune is wonderful, but it was origninally a French Carol, and the English words have always seemed a bit feeble.
This is what I have so far, based loosely on Romans Ch 5 with a touch of Galatians 4:4:
Still was the night
The silent world was waiting
The time was right
And the place was prepared.
Still in its sin
And full of hurt and hating
The world was weak,
But a rescue was dared.
God sent His Son
A baby frail and humble
In peril’s place,
To suffer in our stead.
Oh! Child of hope,
We tremble lest he stumble
In dangerous days.
Oh! Child, in manger bed,
You hold our hope;
Oh! Hope, hope of the world.
Obviously, futher verses to follow.
Presents were unexpected and funny. I had an enormous kipper tie that is held on by an elastic band, a mounted football shirt of Aldershot Town, the club for which I was a mascot during the 1950-51 season and a full set of the Sharpe DVDs. I gave my wife specialty herbal teas, a really expensive jar of lemon curd, and a number of biographies including one of Philipp Bliss, the author of "Man of Sorrows".
We played 3 games of Scrabble, which everybody won except me. We opened a bottle of champagne that had been given to my daughter, the doctor, by a patient, and in between eating (the food was excellent) I worked on a new carol. I have never really liked the words of O Holy Night. The tune is wonderful, but it was origninally a French Carol, and the English words have always seemed a bit feeble.
This is what I have so far, based loosely on Romans Ch 5 with a touch of Galatians 4:4:
Still was the night
The silent world was waiting
The time was right
And the place was prepared.
Still in its sin
And full of hurt and hating
The world was weak,
But a rescue was dared.
God sent His Son
A baby frail and humble
In peril’s place,
To suffer in our stead.
Oh! Child of hope,
We tremble lest he stumble
In dangerous days.
Oh! Child, in manger bed,
You hold our hope;
Oh! Hope, hope of the world.
Obviously, futher verses to follow.
Saturday, December 24, 2005
Publication ethics
I e-mailed Lou Staudt when I saw his paper before it was published and asked him to come to England and talk about it. He seemed pleased to hear from me. We had not met previously but our work had suddenly become complimentary. The prognosis was so different between mutated and unmutated CLL that the question had to be asked whether they were both the same disease. Lou had developed his Lymphochip as a way of looking at which genes were switched and which switched off in a particular tissue. He had already explored the complexity of diffuse large cell lymphoma and it was an obvious move to look at CLL. His results clearly showed that CLL was a single disease very different from any other lymphoma and different also from most types of B cell.
What I didn’t understand was why it took so long to get published. We knew that it had been circulating around high impact journals and it was obviously a very important paper. We began to think that dark forces were preventing its publication.
Publication ethics is a can of worms. Referees are on their honor to say if they have a competing interest. Authors should reveal whether the paper has been published before in part or whole. Editors should not employ chicanery to raise their impact factor. All have sinned and fallen short of the glory of God.
When it was published it was brilliant. It identified a number of genes which separated the mutated and unmutated subset. Most important of these was ZAP-70. This could have been easily missed because it is always switched on in T cells, so unless the T cells had been depleted from the CLL specimens it would not have been found.
Lou couldn’t make the meeting, but Andreas Rosenwald (who is about 7ft tall) came instead and there began a useful collaboration. We began to develop tests for ZAP-70 and by 2002 we had a working flow cytometry test that remains our benchmark assay.
Emili Montserrat’s group in Barcelona developed a very similar test, again ready for the 2002 ASH meeting, and then last year Tom Kipps’ group working with the CLL Consortium developed a single step flow assay that can be used on whole blood.
Guess what? Concordance with VH genes isn’t perfect, and in fact with some assays it is hardly better than CD38.
What I didn’t understand was why it took so long to get published. We knew that it had been circulating around high impact journals and it was obviously a very important paper. We began to think that dark forces were preventing its publication.
Publication ethics is a can of worms. Referees are on their honor to say if they have a competing interest. Authors should reveal whether the paper has been published before in part or whole. Editors should not employ chicanery to raise their impact factor. All have sinned and fallen short of the glory of God.
When it was published it was brilliant. It identified a number of genes which separated the mutated and unmutated subset. Most important of these was ZAP-70. This could have been easily missed because it is always switched on in T cells, so unless the T cells had been depleted from the CLL specimens it would not have been found.
Lou couldn’t make the meeting, but Andreas Rosenwald (who is about 7ft tall) came instead and there began a useful collaboration. We began to develop tests for ZAP-70 and by 2002 we had a working flow cytometry test that remains our benchmark assay.
Emili Montserrat’s group in Barcelona developed a very similar test, again ready for the 2002 ASH meeting, and then last year Tom Kipps’ group working with the CLL Consortium developed a single step flow assay that can be used on whole blood.
Guess what? Concordance with VH genes isn’t perfect, and in fact with some assays it is hardly better than CD38.
Friday, December 23, 2005
In the Bleak Midwinter
I am listening to a delicious version of Christina Rossetti's famous carol from the choir of New College, Oxford. This perhaps is the msot beautiful of all the Chistmas songs. Nearly 30 years ago I learned the tenor part for a Christmas performance. (Nowadays I have to sing bass; I have Le sang froid Anglais - what Noel Coward translated as the Englishman and his usual bloody cold.)
Of course the words are inappropriate for Palestine in September - or whenever it was the wondrous deed was done. No snow; no frosty wind to make any moan; no earth as hard as iron; no water like a stone. No heat in the very sod the saint has printed. No winter's snow to see amidst. No cold winter's night that was so deep. But in truth not many carols have any reference to the sort of Northern European winter conditions that make them difficult to sing in the antipodes. Winds through the olive trees has the sheep on Bethelhem hills knee deep in snow, but generally the snowy Christmas card imagary is avoided.
The difficulty of the bleak midwinter is that in the last line everybody fails to fit all the words in. The "Yet what" has to be sung on the first note. and then it continues, "I can I give Him; give my heart!"
Of course the words are inappropriate for Palestine in September - or whenever it was the wondrous deed was done. No snow; no frosty wind to make any moan; no earth as hard as iron; no water like a stone. No heat in the very sod the saint has printed. No winter's snow to see amidst. No cold winter's night that was so deep. But in truth not many carols have any reference to the sort of Northern European winter conditions that make them difficult to sing in the antipodes. Winds through the olive trees has the sheep on Bethelhem hills knee deep in snow, but generally the snowy Christmas card imagary is avoided.
The difficulty of the bleak midwinter is that in the last line everybody fails to fit all the words in. The "Yet what" has to be sung on the first note. and then it continues, "I can I give Him; give my heart!"
Last minute
It is a tradition in our house to fill stockings for the children. This year our two youngest and unmarried children (aged 25 and 28) will be with us. I have just added final trimmings to the stockings. A 30p card game (Old Maid and Donkey), a 20p lollipop, a chocolate orange and a pencil. All the while playing an absolutely dreadful Christmas CD: Timeless Christmas Classics. Pat Boone, Burl Ives, Beverly Shea and George Hamilton IV. They are anything but timeless. They are frozen in time - about 1957 I think.
CD38 and VH mutations are independent
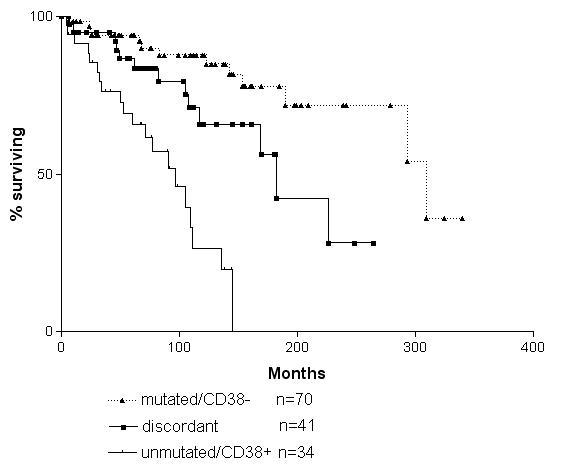
CLLs discordant for VH mutations and CD38 have an intermediate prognosis

CD38 is discordant
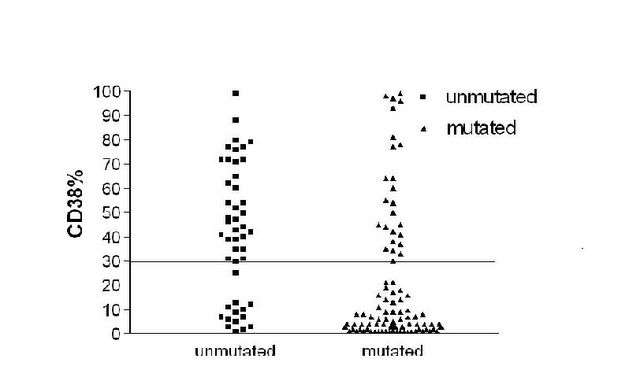
This graph shows the lack of correlation between VH mutations and CD38. About 30% are discordant.
Chiorazzi graph

This was the original graph from the Chiorazzi paper showing that CD38 was a useful prognostic factor
CD38
Somebody recently asked on the ACOR List CD38 and VH genes interalate. Let's first think about CD38. It has been mentioned in well over 1000 articles in the past five years, so what is it?
It is a type II transmembrane glycoprotein, the extracellular domain acting as an ectoenzyme, catalyzing the conversion of NAD+ into nicotinamide, ADP-ribose (ADPR) and cyclic ADPR.
That means almost nothing to most people. Let's just say that is a molecule that lives on and in the cell membrane and it is an enzyme; that is, it catalyzes chemical reactions.
It is found on many types of cells, but we are really interested in its role on B-lymphocytes. It is there from time to time as the B cell differentiates (this is a word we introduced yesterday. It means the changes that happen as the cell grows ups from a baby to maturity). It appears on bone marrow precursor cells, but is lost on mature lymphocytes; on germinal center cells it protects against apoptosis, but on leaving the germinal center, memory cells lack the antigen; on terminally differentiated plasma cells it is one of the few surface antigens present. In chronic lymphocytic leukemia (CLL) expression of CD38 signifies a poor prognosis although it does not correlate precisely with the presence of unmutated immunoglobulin variable region (IgV) genes and it may vary during the course of the disease.
Is it more than a prognostic marker? Dr Malavasi and his colleagues in Turin think that CD38 is involved in signaling through the B-cell receptor (BCR).
When we published out first paper on VH genes as prognostic markers, the accompanying paper from Nick Chiorazzi's group in Long Island confirmed our finding and also suggested that CD38 (which is much easier to measure) could act as a surrogate because it gave the same answer.
When we looked at CD38 in our series it gave a very similar answer, but in idividual cases there was a 30% disparity between CD38 levels and VH gene mutations. Those who were discordant for VH genes and CD38, whether it was CD38 positive and VH mutated or CD38 negative and VH unmutated had an average survival midway between the 8 years and 25 years that we had found for mutated versus unmutated. It was 15 years.
The other surprising thing was that of 40 patients in whom we were to follow for a long time we found that the CD38 level changed. Why this was so is complicated. We know that some cytokines like IL-2 and interferon gamma can alter the level of CD38 present, but we also know that after treatment the cells that remain (those resistant to the drugs) often show more CD38 present
It is a type II transmembrane glycoprotein, the extracellular domain acting as an ectoenzyme, catalyzing the conversion of NAD+ into nicotinamide, ADP-ribose (ADPR) and cyclic ADPR.
That means almost nothing to most people. Let's just say that is a molecule that lives on and in the cell membrane and it is an enzyme; that is, it catalyzes chemical reactions.
It is found on many types of cells, but we are really interested in its role on B-lymphocytes. It is there from time to time as the B cell differentiates (this is a word we introduced yesterday. It means the changes that happen as the cell grows ups from a baby to maturity). It appears on bone marrow precursor cells, but is lost on mature lymphocytes; on germinal center cells it protects against apoptosis, but on leaving the germinal center, memory cells lack the antigen; on terminally differentiated plasma cells it is one of the few surface antigens present. In chronic lymphocytic leukemia (CLL) expression of CD38 signifies a poor prognosis although it does not correlate precisely with the presence of unmutated immunoglobulin variable region (IgV) genes and it may vary during the course of the disease.
Is it more than a prognostic marker? Dr Malavasi and his colleagues in Turin think that CD38 is involved in signaling through the B-cell receptor (BCR).
When we published out first paper on VH genes as prognostic markers, the accompanying paper from Nick Chiorazzi's group in Long Island confirmed our finding and also suggested that CD38 (which is much easier to measure) could act as a surrogate because it gave the same answer.
When we looked at CD38 in our series it gave a very similar answer, but in idividual cases there was a 30% disparity between CD38 levels and VH gene mutations. Those who were discordant for VH genes and CD38, whether it was CD38 positive and VH mutated or CD38 negative and VH unmutated had an average survival midway between the 8 years and 25 years that we had found for mutated versus unmutated. It was 15 years.
The other surprising thing was that of 40 patients in whom we were to follow for a long time we found that the CD38 level changed. Why this was so is complicated. We know that some cytokines like IL-2 and interferon gamma can alter the level of CD38 present, but we also know that after treatment the cells that remain (those resistant to the drugs) often show more CD38 present
VH mutations graph

One picture is worth a thousand words. This survival curve shows the difference between having mutated and unmutated VH genes.
Thursday, December 22, 2005
VH mutations
I well remember a meeting in Italy when VH gene mutations were the hot topic. I was in the camp that believed that CLL was frozen in time as a pre-germinal center tumor, with unmutated VH genes. I guess this was based on no stronger evidence than that CLL cells had IgD molecules on their surface. IgD is an unusual form of immunoglobulin, almost only found as a surface molecule and usually at an early stage of differentiation. Several centers had been sequencing the DNA of the immunoglobulin chains in small numbers of patients. Probably the greatest number had been sequenced by Tom Kipps from San Diego, and as an admirer of Tom, I was plaesed to see that his findings fitted with my prejudices.
Guillaume Dighiero from Paris and Harry Schroeder from the US had assembled all the published case reports of CLLs with VH gene sequences. They found 76 and much to my surprise half of them had mutated VH genes. I scrutinised these cases carefully and was very dubious. Several of the mutated cases had been reported by Nick Chiorazzi from North Shore, Long Island, and had surface IgG rather than IgM+D. This is unusual in CLL and since class switching from M to G is a germinal center task it was natural that such cases should be mutated, another germinal center task. Other cases were clearly not CLL, being CD5 negative and still others had serum paraproteins, an unusual finding in CLL.
The way we got into VH gene sequencing was like this. Martin Glennie had been raising anti-idiotype antibodies against lymphomas as part of our lymphoma therapy protocol. One of these antibodies, 9G4, proved of no value as a therapy because it cross reacted with other antibodies. The patient happened to make a cold agglutinin and Freda Stevenson had the idea that 9G4 might cross-react with all cold aglutinins.
Cold agglutinins had been an interest of mine since I was a juior doctor. I should expplain that cold agglutinins are IgM antibodies that are present in everybody's blood at low concentrations, that bind to red blood cells in the cold and cause them to stick together. In certain circumstances high levels of cold agglutinins are found in the blood - after infection with mycoplasma pneumoniae or EB virus, and in some elderly people where the antibody is monoclonal, and in some lymphomas where it is made by the tumor.
So we set about collecting all the cold agglutinin specimens we could find. We produced cell lines that secreted cold agglutinins. It turned out to be true. All cold agglutinins reacted with 9G4. We then sent the cell lines to America to have the immunoglobulin genes sequenced. This demonstrated that all cold agglutinins used the same VH gene. It was then known as V4-21 but has now been renamed as V4-34. It soon became clear that we could not continue this research with someone doing the sequencing for us so we had to learn to do it ourselves. Doing it by hand is a tedious time-consuming business so I was able to raise some money to buy a machine to automate it.
Freda Stevenson and David Oscier looked at 20 CLLs and sure enough found that both mutated and unmutated V genes were present. The striking finding was that the mutated cases had the del 13q14 chromosomal abnormality while the unmutated one had trisomy 12. This made us suspicious that the more benign cases were mutated, and to test this I collected another 64 cases. Now that we had large numbers, there was no doubt about it; the mutated cases had an average survival of 25 years and the unmutated cases only 8 years. I presented these data in 1997 and 1998 at the British Society for Haematology, the IWCLL and the ASH meetings. Nick Chiorazzi heard the presentation at the IWCLL. He had sequences on 64 cases and correlated their survival with the VH gene status and found exactly the same as we had. He also found that CD38 expression gave very similar results. We decided to publish the papers together as being mutually confirmatory.
Following this paper the value of VH gene mutational status has been confirmed several times by the French, Germans, and Swedes in particular.
Guillaume Dighiero from Paris and Harry Schroeder from the US had assembled all the published case reports of CLLs with VH gene sequences. They found 76 and much to my surprise half of them had mutated VH genes. I scrutinised these cases carefully and was very dubious. Several of the mutated cases had been reported by Nick Chiorazzi from North Shore, Long Island, and had surface IgG rather than IgM+D. This is unusual in CLL and since class switching from M to G is a germinal center task it was natural that such cases should be mutated, another germinal center task. Other cases were clearly not CLL, being CD5 negative and still others had serum paraproteins, an unusual finding in CLL.
The way we got into VH gene sequencing was like this. Martin Glennie had been raising anti-idiotype antibodies against lymphomas as part of our lymphoma therapy protocol. One of these antibodies, 9G4, proved of no value as a therapy because it cross reacted with other antibodies. The patient happened to make a cold agglutinin and Freda Stevenson had the idea that 9G4 might cross-react with all cold aglutinins.
Cold agglutinins had been an interest of mine since I was a juior doctor. I should expplain that cold agglutinins are IgM antibodies that are present in everybody's blood at low concentrations, that bind to red blood cells in the cold and cause them to stick together. In certain circumstances high levels of cold agglutinins are found in the blood - after infection with mycoplasma pneumoniae or EB virus, and in some elderly people where the antibody is monoclonal, and in some lymphomas where it is made by the tumor.
So we set about collecting all the cold agglutinin specimens we could find. We produced cell lines that secreted cold agglutinins. It turned out to be true. All cold agglutinins reacted with 9G4. We then sent the cell lines to America to have the immunoglobulin genes sequenced. This demonstrated that all cold agglutinins used the same VH gene. It was then known as V4-21 but has now been renamed as V4-34. It soon became clear that we could not continue this research with someone doing the sequencing for us so we had to learn to do it ourselves. Doing it by hand is a tedious time-consuming business so I was able to raise some money to buy a machine to automate it.
Freda Stevenson and David Oscier looked at 20 CLLs and sure enough found that both mutated and unmutated V genes were present. The striking finding was that the mutated cases had the del 13q14 chromosomal abnormality while the unmutated one had trisomy 12. This made us suspicious that the more benign cases were mutated, and to test this I collected another 64 cases. Now that we had large numbers, there was no doubt about it; the mutated cases had an average survival of 25 years and the unmutated cases only 8 years. I presented these data in 1997 and 1998 at the British Society for Haematology, the IWCLL and the ASH meetings. Nick Chiorazzi heard the presentation at the IWCLL. He had sequences on 64 cases and correlated their survival with the VH gene status and found exactly the same as we had. He also found that CD38 expression gave very similar results. We decided to publish the papers together as being mutually confirmatory.
Following this paper the value of VH gene mutational status has been confirmed several times by the French, Germans, and Swedes in particular.
Wednesday, December 21, 2005
Frozen in time
I need to say something about differentiation. The whole of life is about stem cells turning into differentiated cells. The most obvious stem cell is the fertilized egg, which has to differentiate into every tissue in the body: liver cells, blood cells, brain cells, skin cells, bowel cells and bladder cells. Within a given tissue there are stem cells committed to different paths of differentiation. For example, in the bone marrow pluripotenial stem cells differentiate in different directions to make lymphocytes, monocytes, erythrocytes, neutrophils, eosinophils, basophils, dendritic cells, platelets, etc.
A true stem cell carries all the possible genes; differentiation involves switching some off and some on. A bone marrow stem cell destined to become a neutrophil goes through stages of differentiation and cell division. It is first a myeloblast, then a promyelocyte, then a myelocyte, then a metamyelocyte then a non-segmented polymorph and finally a neutrophil which struts and frets its hour upon the stage and then is seen no more.
In most cancers, the process of differentiation is interfered with. A cell becomes frozen in time; incapable of differentiating further. In acute myeloblastic leukemia, cells are frozen at the myeloblast stage.
The differentiation of the B lymphocyte was described yesterday, but in different terms. A B lymphocyte becomes committed to make a particular antibody by all that rearrangement of the V, D and J genes. The next stage is to enter the germinal center of the lymph node where somatic mutation takes place, making the antibody the best fit possible for the antigen. From there the B cells either migrates back to the bone marrow to become a plasma cell where it secretes its antibody, or it circulates in the blood as a memory cell.
Throughout the 1990s the great debate was about whether the CLL cell was frozen as a pre-germinal center cell or a post-germinal center cell. There were believers on both sides. Tomorrow I will tell you who won.
A true stem cell carries all the possible genes; differentiation involves switching some off and some on. A bone marrow stem cell destined to become a neutrophil goes through stages of differentiation and cell division. It is first a myeloblast, then a promyelocyte, then a myelocyte, then a metamyelocyte then a non-segmented polymorph and finally a neutrophil which struts and frets its hour upon the stage and then is seen no more.
In most cancers, the process of differentiation is interfered with. A cell becomes frozen in time; incapable of differentiating further. In acute myeloblastic leukemia, cells are frozen at the myeloblast stage.
The differentiation of the B lymphocyte was described yesterday, but in different terms. A B lymphocyte becomes committed to make a particular antibody by all that rearrangement of the V, D and J genes. The next stage is to enter the germinal center of the lymph node where somatic mutation takes place, making the antibody the best fit possible for the antigen. From there the B cells either migrates back to the bone marrow to become a plasma cell where it secretes its antibody, or it circulates in the blood as a memory cell.
Throughout the 1990s the great debate was about whether the CLL cell was frozen as a pre-germinal center cell or a post-germinal center cell. There were believers on both sides. Tomorrow I will tell you who won.
Tuesday, December 20, 2005

The B cell receptor
The B cell receptor is what makes a B cell a B cell. It's main component is the immunoglobulin molecule that it is programmed to make. It's like a label on the surface of the cell that says, "This is what I make."
Back in the 1960s I though that antibodies were made to order, formed around the template of antigen. I well remember the lecture by John Verrier Jones who scotched this idea. He started by explaining how diverse was the immune response. Millions of different antibodies were requred. Yet it was improbably specific. A slight change in shape would mean that the antibody would no longer react with the antigen. Surely every antibody must be made individually; built to the specifications of the antigen.
But No. This is not how nature works. The usual method is to make every possible shape and select from these. Survival of the fittest is standard practice. The objection is that to produce every possible antibody requires more information than is present in the DNA. As John put it, "The information required would take up 44 chromosomes with only 2 left for all the other things the body needs to do."
The answer is mix and match. In the human there are 5 genes that code for the buiness end of the antibody - the end that combines with the antigen. The antibody molecule consists of 4 polypeptide chains (that means 4 chains made up of a sequence of amino acids). There are 2 identical light chains and 2 identical heavy chains. For the heavy chains the chains are Variable (VH) Joining (JH) and Diversity (D), while for the light chain there are Variable (VL) and Joining (JL). In order to obtain variation the chromosomes contain many different copies of these genes, all slightly different. So there are 51 different VH genes, 6 different JH genes and 27 different D genes. Something similar exists for the lightchain. Every time a new lymphocyte is born it selects one of the 51 VH genes, one of the 6 JH genes and one of the 27 D genes. Thus it decides to use only one of the 8262 possible combinations.
It is even more complicated than that. The junction between V and D and D and J is imprecise. It is like my carpentry. When I make a joint, sometimes there is a gap and I have to add filler to fill it; sometimes the fit is too tight and I have to plane a bit off to make the fit. The same is true with the immunoiglobulin genes sometimes the cellneedes to add filler, sometimes to plane a bit off.
To explain how this works we have to understand the DNA code.
DNA is like a spiral staircase. The bannisters are made up from the sugar backbone; the steps from the nucleotide bases. In DNA there are 4 types of nucleotide: Guanine (G), Thymidine (T), Cytosine (C) and Adenine (A). In forming the steps C always pairs with G and A always pairs with T. This makes copying easy because if the two halves of the chain come apart as they do in cell division, each acts as a template for the construction of the complementary chain.
The DNA is translated into protein, which is made up of chains of amino acids. In the DNA a sequence of three nucleotides is called a triplet. There are 64 possible triplets and 20 possible amino acids. Some amino acids are coded for by 4 triplets and some by 2, while one triplet codes for 'start copying' and one for 'stop copying'. Thus GCU codes for alanine, AGC for serine, UGC for cystine, GAC for aspartic acid, CUG for leucine, AUG for methionine etc. When extra filler nucleotides are added it changes the whole sequence.
For example, imagine a sequence of alanines:
GCU GCU GCU GCU GCU GCU GCU
ala ala ala ala ala ala ala
An extra nucleotide insertion does this:
GCU GCU AGC UGC UGC UGC UGC
ala ala ser cys cys cys cys
Removing a nucleotide does this:
GCU GCU GCU CUG CUG CUG CUG
ala ala ala leu leu leu leu
These are called frame shifts: obviously any sequence of nucleotides can be read in any of 3 frames. It means that the number of possibles is now multiplied by 3 up to over 24,000, plus the extra nucleotides are not limited to one or two but may be many: it could be very bad carpentry.
When the light chain possibilities are added in we are up into the millions.
There is even more refinement to come. The usual thing is for antigen to select out the BCR that fits it best and to take it along to the nearest germinal center where antigen and antibody start canoodling. The B cell reacts by dividing and each time it does so it makes a subtle variation in the BCR by a small number of point mutations. These are called somatic mutations. Some of these will make the antibody a dreadful fit for the antigen: such cells are executed by apoptosis. But some will be a better fit, and by such selective breeding the dog with just the right length of tail is produced. Thus the best antibody for any immune response is made.
Back in the 1960s I though that antibodies were made to order, formed around the template of antigen. I well remember the lecture by John Verrier Jones who scotched this idea. He started by explaining how diverse was the immune response. Millions of different antibodies were requred. Yet it was improbably specific. A slight change in shape would mean that the antibody would no longer react with the antigen. Surely every antibody must be made individually; built to the specifications of the antigen.
But No. This is not how nature works. The usual method is to make every possible shape and select from these. Survival of the fittest is standard practice. The objection is that to produce every possible antibody requires more information than is present in the DNA. As John put it, "The information required would take up 44 chromosomes with only 2 left for all the other things the body needs to do."
The answer is mix and match. In the human there are 5 genes that code for the buiness end of the antibody - the end that combines with the antigen. The antibody molecule consists of 4 polypeptide chains (that means 4 chains made up of a sequence of amino acids). There are 2 identical light chains and 2 identical heavy chains. For the heavy chains the chains are Variable (VH) Joining (JH) and Diversity (D), while for the light chain there are Variable (VL) and Joining (JL). In order to obtain variation the chromosomes contain many different copies of these genes, all slightly different. So there are 51 different VH genes, 6 different JH genes and 27 different D genes. Something similar exists for the lightchain. Every time a new lymphocyte is born it selects one of the 51 VH genes, one of the 6 JH genes and one of the 27 D genes. Thus it decides to use only one of the 8262 possible combinations.
It is even more complicated than that. The junction between V and D and D and J is imprecise. It is like my carpentry. When I make a joint, sometimes there is a gap and I have to add filler to fill it; sometimes the fit is too tight and I have to plane a bit off to make the fit. The same is true with the immunoiglobulin genes sometimes the cellneedes to add filler, sometimes to plane a bit off.
To explain how this works we have to understand the DNA code.
DNA is like a spiral staircase. The bannisters are made up from the sugar backbone; the steps from the nucleotide bases. In DNA there are 4 types of nucleotide: Guanine (G), Thymidine (T), Cytosine (C) and Adenine (A). In forming the steps C always pairs with G and A always pairs with T. This makes copying easy because if the two halves of the chain come apart as they do in cell division, each acts as a template for the construction of the complementary chain.
The DNA is translated into protein, which is made up of chains of amino acids. In the DNA a sequence of three nucleotides is called a triplet. There are 64 possible triplets and 20 possible amino acids. Some amino acids are coded for by 4 triplets and some by 2, while one triplet codes for 'start copying' and one for 'stop copying'. Thus GCU codes for alanine, AGC for serine, UGC for cystine, GAC for aspartic acid, CUG for leucine, AUG for methionine etc. When extra filler nucleotides are added it changes the whole sequence.
For example, imagine a sequence of alanines:
GCU GCU GCU GCU GCU GCU GCU
ala ala ala ala ala ala ala
An extra nucleotide insertion does this:
GCU GCU AGC UGC UGC UGC UGC
ala ala ser cys cys cys cys
Removing a nucleotide does this:
GCU GCU GCU CUG CUG CUG CUG
ala ala ala leu leu leu leu
These are called frame shifts: obviously any sequence of nucleotides can be read in any of 3 frames. It means that the number of possibles is now multiplied by 3 up to over 24,000, plus the extra nucleotides are not limited to one or two but may be many: it could be very bad carpentry.
When the light chain possibilities are added in we are up into the millions.
There is even more refinement to come. The usual thing is for antigen to select out the BCR that fits it best and to take it along to the nearest germinal center where antigen and antibody start canoodling. The B cell reacts by dividing and each time it does so it makes a subtle variation in the BCR by a small number of point mutations. These are called somatic mutations. Some of these will make the antibody a dreadful fit for the antigen: such cells are executed by apoptosis. But some will be a better fit, and by such selective breeding the dog with just the right length of tail is produced. Thus the best antibody for any immune response is made.
Monday, December 19, 2005
Frogs
A hugely funny cartoon in today's Times.
In the first panel Tony Blair is confronted by a frog with the face of Jacques Chirac. "Ah well," he says, "Sometimes you have to kiss the frog".
In the second panel he grimaces as he kisses the frog.
In the third panel there are flashes and stars (including the EU circle of stars. Obviously magic is taking place.
In the fourth panel Tony Blair has been turned into a small frog.
My favorite version of the frog joke, which I told at the retirement party of Jacques-Louis Binet goes like this:
A frog hopped up to an elderly professor, "If you kiss me I will turn into a beautiful princess."
The professor ignores the frog and walks by. The frog hops faster to catch up. "If you kiss me I will turn into a beautiful princess and I'm all yours,"
The professor still ignores the frog and walks on. The frog redoubles its efforts and catches up again. "If you kiss me I will turn into a beautiful princess and I'm all yours to do what you like with."
At this the professor bends down, scoops up the frog and puts it in his pocket. From the pocket comes a mufled protest, "Hey! You're supposed to kiss me."
The professor pulls out the frog and says to it, "At my age, a talking frog is worth more."
To understand the EU you have to realise what happened after the first world war. At the Treaty of Versailles the French exacted such heavy war reparations from the Germans that they bankrupted them. This led directly to the rise of Hitler and the second world war. After this the European powers decided that they needed a treaty to prevent another Franco-German war. The Common Market was a means by which the French exacted war reparations from the Germans without precipitating a third world war. The financial arangements involved the German subsidising inefficient French farmers in perpetuity. But how were the Germans to pay? The simple answer is the Marshall plan. So after the Americans rescued France from the German oppressor they ended up paying the German war reparations to the French. In order to avoid Britain getting caught in the same trap Mrs Thatcher got her rebate.
In the first panel Tony Blair is confronted by a frog with the face of Jacques Chirac. "Ah well," he says, "Sometimes you have to kiss the frog".
In the second panel he grimaces as he kisses the frog.
In the third panel there are flashes and stars (including the EU circle of stars. Obviously magic is taking place.
In the fourth panel Tony Blair has been turned into a small frog.
My favorite version of the frog joke, which I told at the retirement party of Jacques-Louis Binet goes like this:
A frog hopped up to an elderly professor, "If you kiss me I will turn into a beautiful princess."
The professor ignores the frog and walks by. The frog hops faster to catch up. "If you kiss me I will turn into a beautiful princess and I'm all yours,"
The professor still ignores the frog and walks on. The frog redoubles its efforts and catches up again. "If you kiss me I will turn into a beautiful princess and I'm all yours to do what you like with."
At this the professor bends down, scoops up the frog and puts it in his pocket. From the pocket comes a mufled protest, "Hey! You're supposed to kiss me."
The professor pulls out the frog and says to it, "At my age, a talking frog is worth more."
To understand the EU you have to realise what happened after the first world war. At the Treaty of Versailles the French exacted such heavy war reparations from the Germans that they bankrupted them. This led directly to the rise of Hitler and the second world war. After this the European powers decided that they needed a treaty to prevent another Franco-German war. The Common Market was a means by which the French exacted war reparations from the Germans without precipitating a third world war. The financial arangements involved the German subsidising inefficient French farmers in perpetuity. But how were the Germans to pay? The simple answer is the Marshall plan. So after the Americans rescued France from the German oppressor they ended up paying the German war reparations to the French. In order to avoid Britain getting caught in the same trap Mrs Thatcher got her rebate.
Natural history of CLL
Natural history is a bit of medical jargon. It means what happens if the disease is left to itself without treatment. For CLL this is very variable.
Some people who are diagnosed with CLL continue to have a raised white cell count, but never develop enlarged lymph nodes or spleen; never develop anemia, neutropenia or thrombocytopenia; never get ill. They die at 105 of something else.
Some people already have large lymph nodes that get bigger, a large spleen that becomes enormous; are anemic making them short of breath, neutropenic so that they catch pneumonia and are thrombocytopenic and bruising easily or bleeding uncontrollably.
And in between almost every variation occurs.
Lymph node enlargement is mainly very superficial. In the neck, groin and armpits the nodes are easlily felt. They tend to be multiple, not painful or tender, quite soft and rubbery and of sizes varying from a small pea to a quail's egg. (How many have seen a quail's egg? If you saw the TV production of Brideshead Revisited you will remember that Sebastian served a huge plate of quail's eggs in his rooms at Oxford.
The spleen enlarges from under the left ribcage. I have only once seen it go past the umbillicus.
The anemia is normochromic, normocytic. That means the red cells are of a normal color and normal size. Severe anemia is unusual and makes one think of autoimmunity.
Autoimmune hemolytic anemia (AIHA) occurs in between 10 and 20% of cases (depending on which paper you read). CLL is actually the commonest known cause of AIHA, acounting for about 25% of cases. An antibody is produced directed against the patient's own red blood cells. It is not made by the CLL cells themselves but by the residual normal B cells. The antibody latches on to the red cells and fixes complement. Complement is a cascade of blood proteins that react one with another from C1 to C9. C9 provides the killer blow and punches a hole in the red cell. However, in AIHA realted to CLL the activation of complement seldom goes beyond C3. C3 makes red cells more attractive to macrophages and as the blood circulates through the spleen the red cells get snaffled by the macrophages there.
Immune thrombocytopenic purpura (ITP) occurs in between 1 and 2% of cases. Again it is cause by an autoantibody. Platelets coated by antibody also get snaffled by the spleen.
Pure red cell aplasia may be an autoimmune disease directed against the red cell precursors so that no red cells are made by the bone marrow. However, it may be that the mere presence of large numbers of CLL cells in the bone marrow is enough to inhibit red cells from growing there.
Most patients with CLL get immunodeficient. The most obvious manifestation of this is the fall in the serum imunoglobulins. The fall is greatest in those who have had it the longest and in the greatest bulk. Patients suffer from sinus infections and pneumonia. Between a quarter and a third get shingles.
Some people who are diagnosed with CLL continue to have a raised white cell count, but never develop enlarged lymph nodes or spleen; never develop anemia, neutropenia or thrombocytopenia; never get ill. They die at 105 of something else.
Some people already have large lymph nodes that get bigger, a large spleen that becomes enormous; are anemic making them short of breath, neutropenic so that they catch pneumonia and are thrombocytopenic and bruising easily or bleeding uncontrollably.
And in between almost every variation occurs.
Lymph node enlargement is mainly very superficial. In the neck, groin and armpits the nodes are easlily felt. They tend to be multiple, not painful or tender, quite soft and rubbery and of sizes varying from a small pea to a quail's egg. (How many have seen a quail's egg? If you saw the TV production of Brideshead Revisited you will remember that Sebastian served a huge plate of quail's eggs in his rooms at Oxford.
The spleen enlarges from under the left ribcage. I have only once seen it go past the umbillicus.
The anemia is normochromic, normocytic. That means the red cells are of a normal color and normal size. Severe anemia is unusual and makes one think of autoimmunity.
Autoimmune hemolytic anemia (AIHA) occurs in between 10 and 20% of cases (depending on which paper you read). CLL is actually the commonest known cause of AIHA, acounting for about 25% of cases. An antibody is produced directed against the patient's own red blood cells. It is not made by the CLL cells themselves but by the residual normal B cells. The antibody latches on to the red cells and fixes complement. Complement is a cascade of blood proteins that react one with another from C1 to C9. C9 provides the killer blow and punches a hole in the red cell. However, in AIHA realted to CLL the activation of complement seldom goes beyond C3. C3 makes red cells more attractive to macrophages and as the blood circulates through the spleen the red cells get snaffled by the macrophages there.
Immune thrombocytopenic purpura (ITP) occurs in between 1 and 2% of cases. Again it is cause by an autoantibody. Platelets coated by antibody also get snaffled by the spleen.
Pure red cell aplasia may be an autoimmune disease directed against the red cell precursors so that no red cells are made by the bone marrow. However, it may be that the mere presence of large numbers of CLL cells in the bone marrow is enough to inhibit red cells from growing there.
Most patients with CLL get immunodeficient. The most obvious manifestation of this is the fall in the serum imunoglobulins. The fall is greatest in those who have had it the longest and in the greatest bulk. Patients suffer from sinus infections and pneumonia. Between a quarter and a third get shingles.
Sunday, December 18, 2005
One week to Christmas
One week to Christmas. Still no snow. It was very cold this morning, frost on the ground, ice on the windows, but by this evening it is muggy and damp again.
Carols this morning in church. New carols. Graham Kendrick carols. I am working my way through my 29 CDs of Christmas songs and carols. From "A lonely pup in a Christmas shop" to Poulenc and Britten. I love them all. My favorites? O Holy Night. Driving home for Christmas. Hark the Herald Angels Sing. The Pogues.
I think it was "Home Alone" that started the extravagent house decoration in the UK. It is a pleasure to drive around the streets at night trying to find the house decorated in the most extreme bad taste. I don't look down on them. They give me joy. I rejoice in the exuberance of it all.
Carols this morning in church. New carols. Graham Kendrick carols. I am working my way through my 29 CDs of Christmas songs and carols. From "A lonely pup in a Christmas shop" to Poulenc and Britten. I love them all. My favorites? O Holy Night. Driving home for Christmas. Hark the Herald Angels Sing. The Pogues.
I think it was "Home Alone" that started the extravagent house decoration in the UK. It is a pleasure to drive around the streets at night trying to find the house decorated in the most extreme bad taste. I don't look down on them. They give me joy. I rejoice in the exuberance of it all.
Saturday, December 17, 2005
Immunophenotyping
All these CD numbers are confusing. It has nothing to do with compact discs. CD stands for clusters of differentiation.
In the 1980s laboratories began to make monoclonal antibodies against blood cells. At that time nobody had any idea how complex was the surface of the lymphocyte. It soon became clear that some antibodies reacted with some lymphocytes and some reacted with others. It was very confusing.
We had known about T and B lymphocytes since the early 1970s. B lymphocytes had immunoglobulin molecules on their surface. T lymphocytes bound tightly to sheep red blood cells. Why anybody should have mixed lymphocytes with sheep blood cells seems a mystery now, but at the time a small industry came into being mixing blood cells from various types of animals with human lymphocytes. Scientists were raiding zoos for the most exotic species. I remember that we kept an ox at a local farm to provide us with red cells for certain experiments. CLL was strange; it was certainly a B cell since it had immunoglobulin on its surface (though only about 10% as much as other B cells) but it also reacted with red blood cells, not from the sheep but from the mouse.
To solve the mystery workshops were held where people with monoclonal antibodies took them along and reacted them with various populations of white cells. Antibodies that seemed to have similar reactivity were placed in a particular "cluster of differentiation" and given a CD number. Where there were not enough different antibodies of a particular type, a provisional "workshop" number was given. Thus the antibody called B1 by the Boston workers became CD20 because there were several similar antibodies. There were several anti-B cell antibodies recognised at that time and they were given successive numbers: CD19, CD20, CD21, CD22, CD23 and CD24. In the same way the earliest anti-T cell antibodies were given the numbers CD1 through to CD8. Campath was unique; it was given the number CDw52 and only later was it allowed to drop the w.
The eighth workshop was held this year and we are now up to 239 CD numbers. Some have suggested that there may be as many as 5000 different proteins on the surface of lymphocytes so who knows where the process will end?
Immunophenotyping is the process by which a cell can be identified by listing the different antibodies that react with it. It used to be done by fluorescence microscopy. A cell was reacted with an antibody that had been labelled with a fluorescent dye. Under the microscope the dye was excited with ultra-violet light to make it glow bright green, and so label the cells that it reacted with. These days it is all done much more quickly, accurately, sensitively and quantitatively with a flow cytometer.
It is not necessary to list all the antibodies that a type of cell reacts with, only the ones that give it a distinct pattern. For CLL the pattern is CD5, CD19, CD20, CD23 positive, weakly positive for surface immunoglobulin and CD79b, and negative for FMC7.
CD5 is expressed on all mature T cells, but not normally on B cells. Its presence on CLL cells is therefore unexpected. It is also present on the cells of mantle cell lymphoma and these two conditions are frequently confused. I shall dicuss this problem in a later blog. There is also a population of normal CD5 positive B cells. In the mouse these live in the peritoneal cavity and are rather special. They are known as B1 cells. In the human CD5 positive cells are present in the follicular mantle of the lymph node and in cord blood. In tissue culture CD5 negative B cells can be induced to express CD5 after certain types of activation. The question of whether CLL is a tumor of B1 cells or whether CD5 is simply an activation marker on CLL is controversial. I will write about it on another occasion.
CD19 is a marker present on all B cells and in some circumstances is involved in signaling through the surface immunoglobulin.
CD20 is present only on B cells. On CLL cells it is more weakly expressed than on other B cells. It is now clear that FMC7 recognises part of the CD20 molecule, but this part is hidden by lipid in most CLL cells so that FMC7 is usually negative in CLL and the CD20 is weaker than normal.
CD23 is one of the receptors for IgE. It is usually undetectable in B cells except for CLL, but it is also present on monocytes and eosinophils.
CD79b is one of the immunoglobulin associated molecules. It is through this molecule that stimulation of the surgace immunoglobulin sends a signal to the inside of the cell.
Immunophenotyping has enabled us to recognise CLL cells when there are only a few present. It has therefore reduced the threshold for diagnosis of CLL from 15,000 lymphocytes to 5000. It has helped us to distinguish CLL from several imposters including splenic lymphoma with villous lymphocytes, mantle cell lymphoma and marginal zone lymphoma.
In the 1980s laboratories began to make monoclonal antibodies against blood cells. At that time nobody had any idea how complex was the surface of the lymphocyte. It soon became clear that some antibodies reacted with some lymphocytes and some reacted with others. It was very confusing.
We had known about T and B lymphocytes since the early 1970s. B lymphocytes had immunoglobulin molecules on their surface. T lymphocytes bound tightly to sheep red blood cells. Why anybody should have mixed lymphocytes with sheep blood cells seems a mystery now, but at the time a small industry came into being mixing blood cells from various types of animals with human lymphocytes. Scientists were raiding zoos for the most exotic species. I remember that we kept an ox at a local farm to provide us with red cells for certain experiments. CLL was strange; it was certainly a B cell since it had immunoglobulin on its surface (though only about 10% as much as other B cells) but it also reacted with red blood cells, not from the sheep but from the mouse.
To solve the mystery workshops were held where people with monoclonal antibodies took them along and reacted them with various populations of white cells. Antibodies that seemed to have similar reactivity were placed in a particular "cluster of differentiation" and given a CD number. Where there were not enough different antibodies of a particular type, a provisional "workshop" number was given. Thus the antibody called B1 by the Boston workers became CD20 because there were several similar antibodies. There were several anti-B cell antibodies recognised at that time and they were given successive numbers: CD19, CD20, CD21, CD22, CD23 and CD24. In the same way the earliest anti-T cell antibodies were given the numbers CD1 through to CD8. Campath was unique; it was given the number CDw52 and only later was it allowed to drop the w.
The eighth workshop was held this year and we are now up to 239 CD numbers. Some have suggested that there may be as many as 5000 different proteins on the surface of lymphocytes so who knows where the process will end?
Immunophenotyping is the process by which a cell can be identified by listing the different antibodies that react with it. It used to be done by fluorescence microscopy. A cell was reacted with an antibody that had been labelled with a fluorescent dye. Under the microscope the dye was excited with ultra-violet light to make it glow bright green, and so label the cells that it reacted with. These days it is all done much more quickly, accurately, sensitively and quantitatively with a flow cytometer.
It is not necessary to list all the antibodies that a type of cell reacts with, only the ones that give it a distinct pattern. For CLL the pattern is CD5, CD19, CD20, CD23 positive, weakly positive for surface immunoglobulin and CD79b, and negative for FMC7.
CD5 is expressed on all mature T cells, but not normally on B cells. Its presence on CLL cells is therefore unexpected. It is also present on the cells of mantle cell lymphoma and these two conditions are frequently confused. I shall dicuss this problem in a later blog. There is also a population of normal CD5 positive B cells. In the mouse these live in the peritoneal cavity and are rather special. They are known as B1 cells. In the human CD5 positive cells are present in the follicular mantle of the lymph node and in cord blood. In tissue culture CD5 negative B cells can be induced to express CD5 after certain types of activation. The question of whether CLL is a tumor of B1 cells or whether CD5 is simply an activation marker on CLL is controversial. I will write about it on another occasion.
CD19 is a marker present on all B cells and in some circumstances is involved in signaling through the surface immunoglobulin.
CD20 is present only on B cells. On CLL cells it is more weakly expressed than on other B cells. It is now clear that FMC7 recognises part of the CD20 molecule, but this part is hidden by lipid in most CLL cells so that FMC7 is usually negative in CLL and the CD20 is weaker than normal.
CD23 is one of the receptors for IgE. It is usually undetectable in B cells except for CLL, but it is also present on monocytes and eosinophils.
CD79b is one of the immunoglobulin associated molecules. It is through this molecule that stimulation of the surgace immunoglobulin sends a signal to the inside of the cell.
Immunophenotyping has enabled us to recognise CLL cells when there are only a few present. It has therefore reduced the threshold for diagnosis of CLL from 15,000 lymphocytes to 5000. It has helped us to distinguish CLL from several imposters including splenic lymphoma with villous lymphocytes, mantle cell lymphoma and marginal zone lymphoma.
Friday, December 16, 2005
Staging
For the past 30 years CLL has been staged by one of two systems devised by two of the great researchers in CLL. Kanti Rai operating in Long Island and Jacques-Louis Binet practising in Paris have been guiding lights for generations of hematologists.
Both staging systems give an estimate of how advanced the disease is based on simple measurements available to everybody.
The Rai system has 5 groups:
Stage 0: Lymphocytosis only.
Stage I: Lymphocytosis plus enlarged lymph nodes
Stage II: Lymphocytosis plus enlarged spleen or liver.
Stage III: Lymphocytosis plus anemia
Stage IV: Lymphocytosis plus thrombocytopenia.
Anemia is defined as an Hb less than 11 g/dL and thrombocytopenia as a platelet count less than 100,000 /cu mm.
Later, stages I and II were combined to be called intermediate-risk and III and IV combined to be called high-risk.
The Binet system has only 3 groups:
Stage A: Lymphocytosis and fewer than 3 lymph node areas involved
Stage B: Lymphocytosis and 3 or more lymph node areas involved
Stage C: Lymphocytosis and anemia or thrombocytopenia or both.
"Lymph node areas" refer to enlargement of lymph nodes in cervical (neck), axillary (armpit) or inguinal (groin) areas or enlargement of spleen or liver.
Anemia for Binet has to be more severe than Rai: Hb less than 10 g/dL, but thrombocytopenia is the same, platelet count less than 100,000 per cu mm.
These staging systems have served us very well, though they have some drawbacks. It has to be emphasized that anemia or thrombocytopenia must be caused by bone marrow infiltration to move a patient into advanced stage disease. Hemolytic anemia or simple iron deficiency don't count and nor does ITP (I'll explain later).
It is important to note that the stage is just a snapshot for how things are now. If a patient is stage C or III or IV the disease is advanced and the prognosis is likely to be poor, but stage A or 0 or I or II disease may be advanced stage in 6 months or 5 years or never. Since the vast majority of patients have early stage disease when diagnosed the future cannot be predicted by staging. It is also important to recognise that a low stage after treatment does not carry the same implications as a low stage before treatment. Stage 0 disease has an average survival of 10 years or more. If you were stage III and become stage 0 after treatment with FCR it doesn't mean that you now have an average survival of more than 10 years. This is especially the case when you are moved out of stage III by treatment with erythropoietin, which raises the Hb without doing anything to the disease. It just makes the bone marrow try harder. You still have the same prognosis as everybody else with stage III.
Another problem is that the normal Hb is higher for men than women - it doesn't have so far to fall before a woman enters stage III or C. This may account for why men do worse than women with this disease. Advanced stage men are already worse off than advanced stage women.
Cropping up a lot this year has been staging by CT scan. All the prognostic information associated with Rai and Binet staging is dependent on clinical staging. To call a patients stage II because a spleen is a bit enlarged on CT scan plays the patient false. A few retroperitoneal lymph nodes do not make a patient stage I. It is only what can be felt with the hand or seen on a blood count that matters. Kanti Rai himself this year said that if CT scans were to be taken into account then all the data on the natural history of CLL accumulated over the past 30 years will have to be jettisoned.
The French group later defined a type of smoldering CLL: Lymphocytosis only with a lymphocyte count no higher than 30,000 per cu mm and an Hb greater or equal to 12 g/dL. They called this group stage A' and other stage A patients stage A''.
The Spanish group also had a version of smoldering leukemia that was a bit more stringent. They required a lymphocyte count no greater than 30,000 per cu mm, and Hb greater or equal to 13 g/dL, non-diffuse bone marrow histology and a lymphocyte doubling time greater than 12 months.
The last thing I want to say about staging is to beware of the Ann Arbor staging method. This is for lymphomas not CLL. Anybody who has bone marrow involvement is stage IV by Ann Arbor. Everybody with CLL has bone marrow involvement, even those with Rai stage 0. Since stage IV is regarded as an indication for treatment, many people have been treated inappropriately.
Both staging systems give an estimate of how advanced the disease is based on simple measurements available to everybody.
The Rai system has 5 groups:
Stage 0: Lymphocytosis only.
Stage I: Lymphocytosis plus enlarged lymph nodes
Stage II: Lymphocytosis plus enlarged spleen or liver.
Stage III: Lymphocytosis plus anemia
Stage IV: Lymphocytosis plus thrombocytopenia.
Anemia is defined as an Hb less than 11 g/dL and thrombocytopenia as a platelet count less than 100,000 /cu mm.
Later, stages I and II were combined to be called intermediate-risk and III and IV combined to be called high-risk.
The Binet system has only 3 groups:
Stage A: Lymphocytosis and fewer than 3 lymph node areas involved
Stage B: Lymphocytosis and 3 or more lymph node areas involved
Stage C: Lymphocytosis and anemia or thrombocytopenia or both.
"Lymph node areas" refer to enlargement of lymph nodes in cervical (neck), axillary (armpit) or inguinal (groin) areas or enlargement of spleen or liver.
Anemia for Binet has to be more severe than Rai: Hb less than 10 g/dL, but thrombocytopenia is the same, platelet count less than 100,000 per cu mm.
These staging systems have served us very well, though they have some drawbacks. It has to be emphasized that anemia or thrombocytopenia must be caused by bone marrow infiltration to move a patient into advanced stage disease. Hemolytic anemia or simple iron deficiency don't count and nor does ITP (I'll explain later).
It is important to note that the stage is just a snapshot for how things are now. If a patient is stage C or III or IV the disease is advanced and the prognosis is likely to be poor, but stage A or 0 or I or II disease may be advanced stage in 6 months or 5 years or never. Since the vast majority of patients have early stage disease when diagnosed the future cannot be predicted by staging. It is also important to recognise that a low stage after treatment does not carry the same implications as a low stage before treatment. Stage 0 disease has an average survival of 10 years or more. If you were stage III and become stage 0 after treatment with FCR it doesn't mean that you now have an average survival of more than 10 years. This is especially the case when you are moved out of stage III by treatment with erythropoietin, which raises the Hb without doing anything to the disease. It just makes the bone marrow try harder. You still have the same prognosis as everybody else with stage III.
Another problem is that the normal Hb is higher for men than women - it doesn't have so far to fall before a woman enters stage III or C. This may account for why men do worse than women with this disease. Advanced stage men are already worse off than advanced stage women.
Cropping up a lot this year has been staging by CT scan. All the prognostic information associated with Rai and Binet staging is dependent on clinical staging. To call a patients stage II because a spleen is a bit enlarged on CT scan plays the patient false. A few retroperitoneal lymph nodes do not make a patient stage I. It is only what can be felt with the hand or seen on a blood count that matters. Kanti Rai himself this year said that if CT scans were to be taken into account then all the data on the natural history of CLL accumulated over the past 30 years will have to be jettisoned.
The French group later defined a type of smoldering CLL: Lymphocytosis only with a lymphocyte count no higher than 30,000 per cu mm and an Hb greater or equal to 12 g/dL. They called this group stage A' and other stage A patients stage A''.
The Spanish group also had a version of smoldering leukemia that was a bit more stringent. They required a lymphocyte count no greater than 30,000 per cu mm, and Hb greater or equal to 13 g/dL, non-diffuse bone marrow histology and a lymphocyte doubling time greater than 12 months.
The last thing I want to say about staging is to beware of the Ann Arbor staging method. This is for lymphomas not CLL. Anybody who has bone marrow involvement is stage IV by Ann Arbor. Everybody with CLL has bone marrow involvement, even those with Rai stage 0. Since stage IV is regarded as an indication for treatment, many people have been treated inappropriately.
Thursday, December 15, 2005
Christmas Carol
Are you ready for Christmas? This year I am early. I have finished my Christmas shopping and wrapped all the presents. I have filled stockings for my children (aged 25 and 28). I have decorated the Christmas tree. My wife has ordered the turkey and baked the Christmas cake.
At various times over the holiday we shall have visits from all four children and all five grandchildren. We shall visit my mother and my father-in-law and see all my siblings.
I doubt it will snow. It seldom does in Bournemouth. I hope it will be crisp and sunny - if not it will be warm, wet and windy. That's how the weather alternates in this part of the world.
I have seen an argument that Jesus Christ was born on 25th September in 6BC. It is calculated on when Zechariah, the father of John the Baptist, took his turn in serving in the Temple and the fact that Herod the Great died in 4BC. The early church was given to syncretism and simply adopted a pagan midwinter festival.
I used to scoff at the artifice of Christmas. Jolly old Santa has escaped from a Coca-Cola advert. The Christmas tree was instituted by Prince Albert. Christmas Cards were simply a piece of Victorian public relations. Oliver Cromwell abolished Christmas as a pagan rite.
But now I realise that Christmas is very special. It may be sentimental but so what? The 25th of December is not the date of Christ's birthday, but as a symbol it is very powerful. It is the darkest time of the year when the night is longest. As the Bible says, "... at just the right time ... while we were still in our sin God sent his son".
This is my Christmas Carol. It may be sung to the tune of the "Ode to Joy" from Beethoven's 9th symphony.
In the silence and the stillness
Of a dark Judean Dawn
In a moment; in a manger
God is as a baby born.
See him stoop from heavenly splendour
See him set his glory by.
God the maker of the mountains
Born a man; for men to die.
In the counsel of the Godhead
Was conceived a mighty plan.
In his justice, in his mercy,
God should die instead of Man.
O what grace, what condescension!
Through the world his great love tell!
Born a baby, born to save us,
Jesus our Immanuel.
See how shepherds come and worship!
Wise men gather from afar,
Angels fill the sky with brightness
Far outshining moon or star.
Let the whole creation praise him!
Loud with adoration sing!
Worship now our wondrous saviour,
Jesus our redeemer king.
At various times over the holiday we shall have visits from all four children and all five grandchildren. We shall visit my mother and my father-in-law and see all my siblings.
I doubt it will snow. It seldom does in Bournemouth. I hope it will be crisp and sunny - if not it will be warm, wet and windy. That's how the weather alternates in this part of the world.
I have seen an argument that Jesus Christ was born on 25th September in 6BC. It is calculated on when Zechariah, the father of John the Baptist, took his turn in serving in the Temple and the fact that Herod the Great died in 4BC. The early church was given to syncretism and simply adopted a pagan midwinter festival.
I used to scoff at the artifice of Christmas. Jolly old Santa has escaped from a Coca-Cola advert. The Christmas tree was instituted by Prince Albert. Christmas Cards were simply a piece of Victorian public relations. Oliver Cromwell abolished Christmas as a pagan rite.
But now I realise that Christmas is very special. It may be sentimental but so what? The 25th of December is not the date of Christ's birthday, but as a symbol it is very powerful. It is the darkest time of the year when the night is longest. As the Bible says, "... at just the right time ... while we were still in our sin God sent his son".
This is my Christmas Carol. It may be sung to the tune of the "Ode to Joy" from Beethoven's 9th symphony.
In the silence and the stillness
Of a dark Judean Dawn
In a moment; in a manger
God is as a baby born.
See him stoop from heavenly splendour
See him set his glory by.
God the maker of the mountains
Born a man; for men to die.
In the counsel of the Godhead
Was conceived a mighty plan.
In his justice, in his mercy,
God should die instead of Man.
O what grace, what condescension!
Through the world his great love tell!
Born a baby, born to save us,
Jesus our Immanuel.
See how shepherds come and worship!
Wise men gather from afar,
Angels fill the sky with brightness
Far outshining moon or star.
Let the whole creation praise him!
Loud with adoration sing!
Worship now our wondrous saviour,
Jesus our redeemer king.
Wednesday, December 14, 2005

This is me
For those who want to know what I look like; this is me. My mother tells me I haven't changed since I was 18 months old. Certainly my hair is the same color and just as fly-away
How do you know if you have CLL?
December 14th 2005
Three-quarters of all cases of CLL are discovered by accident. The patient has a blood test for some other reason or, these days, for no very good reason at all.
Everybody thinks that CLL is getting commoner and they blame the increased use of pesticides in farming, or the nuclear testing in the sixties or diesel fumes or global warming or mobile phone masts or sunspot activity or Chernobyl or whatever is the current scare story in the newspapers. I suspect that it is not getting commoner at all - it is simply that we are recognising cases that we never used to find.
When I was young, everyday I looked at all the blood tests performed on everyone from a population of half a million. On a good day there were 300 blood counts. Thirty years later my responsibilities had diminished; I only looked at the blood counts from a quarter of a million. But most days there were over 700 tests. Thirty years ago you needed to have a lymphocyte count in the blood of 15,000 befor you could diagnose CLL. Today it is only 5000. Thirty years ago we had just discovered that CLL was a B cell neoplasm. Today we can distinguish several types of B cell neoplasm in the blood. We now know that 3.5% of people over 40 have a population of monoclonal B cells indistinguishable from CLL cells in their blood. The harder you look; the more you find.
The 25% in whom the CLL is not found by chance have a variety of symptoms and signs. (A symptom is something the patient feels; a sign is a physical abnormality the doctor finds.) The commonest finding is an enlarged lymph node.
Lymphocytes circulate through the body like other blood cells. For many their journey is just like other blood cells: out in the arteries, home in the veins. They have another circulation, though. The little thin-walled blood vessels that join the arteries to the veins are called capillaries. Lymphocytes can squeeze out through the walls of the capillaries and enter the tissues. From here they make their way home via the lymphatics, which are like veins for white blood cells. Lymph nodes are like stations on the lymphatic railway. They are normally small - about the size of a split pea - and organised. The job of lymphocytes is to fight against foreign invaders, and the lymph node is where they get their training and their orders before going into battle.
Lymph nodes enlarge during an immune response - you often feel 'glands' in your neck when you have a sore throat. In CLL they enlarge because the cells that are growing in an uncontrolled way normally live in the lymph nodes. In CLL we look for lymph nodes in particular places: the neck - we call these cervical nodes; the armpit - axillary nodes; and the groin - inguinal nodes. But there are lymph nodes all over the body and in CLL any lymph node can enlarge. Mostly they can't be felt, but there are some characteristic lymph nodes that are sometimes felt: submental nodes are under the point of the chin and epitrochlear nodes by the elbow. Lymph nodes at the back of the tummy - retroperitoneal nodes - cannot be felt with the hand but may be shown up by CT scan or ultrasound. Nodes in the chest also have to be visualized by some sort of imaging technique.
The spleen is about the size of a fist and is found deep under the ribs on the left of the tummy. It enlarges in CLL, but it has to get to three times its normal size before it can be felt. As it gets bigger it grows across the tummy towards the right groin. It seldom gets as big as this in CLL but it can grow this large in some kinds of hematological cancers. When it is examined by CT scan in CLL it is almost always found to be enlarged and patients think that they must therefore have advanced stage disease. This is misleading. As far as CLL is concerned a spleen is only considered to be enlarged if the doctor can feel it with his/her hand.
The spleen can be though of as a large lymph node attached to the blood circulation. But it also has other roles. It is the last resting place for dying red cells and platelets. When it enlarges it is apt to take out red cells and platelets before they are ready to go. This is known as hypersplenism and it results in anemia and thrombocytopenia.
Anemia means a reduction of the amount of hemoglobin in the blood (the abreviation in Hb not Hg; Hg is the abreviation for Mercury). Men and women have different normal ranges for Hb. For men it is 13.5 - 17 g/dL, for women 11.5 - 15 g/dL. Anemia cause tiredness, breathlessness, weakness, pale skin and mucous membranes and, in severe cases, the features of heart failure; but in many cases mild anemia has no signs or symptoms. Anemia in CLL is caused by suppression of the bone marrow by the infiltrating lymphocytes, or by hemolysis - the distruction of red cells by an antibody, or by hypersplenism. Of course CLL patients can become anemic for reasons unrelated to CLL, such as iron deficiency or pernicious anemia.
Thrombocytopenia means a reduction of the platelets in the blood. The normal range for platelets is 150,000 to 400,000 per cubic millimeter. The features of thrombocytopenia are bleeding and bruising. Platelets are there to plug holes in broken blood vessels and to help the blood to clot. Bleeding into the skin is called purpura and there are two types: petechiae are tiny pinhead sized blood spots; echymoses are anything larger. Thrombocytopenia can have the same causes as anemia: marrow infiltration, immune destruction and hypersplenism.
Patients may also suffer from so-called B symptoms. B symptoms come from Hodgkin's disease doctors who used them to decide whether to treat with chemotherapy or radiotherapy and they have been imported into CLL theology where they don't necessarily fit precisely. There are three types of B symptom: fever, weight loss and night sweats. Some doctors and most patients think that fatigue ahould also be a B symptom in CLL. I shall write about fatigue on another occasion.
Patients with CLL are more susceptible to infections that normal - they have an immunodeficiency. I will write about this in detail later.
To summarize: Most patients with CLL are discovered because they have a blood test for some other reason. If patients have signs or symptoms, the commonest is enlarged lymph nodes, but the spleen (or more rarely the liver) may also be enlarged. In the most severe cases there may be anemia or thrombocytopenia. Some patients have B symptoms and all have a degree of immunodeficiency.
Three-quarters of all cases of CLL are discovered by accident. The patient has a blood test for some other reason or, these days, for no very good reason at all.
Everybody thinks that CLL is getting commoner and they blame the increased use of pesticides in farming, or the nuclear testing in the sixties or diesel fumes or global warming or mobile phone masts or sunspot activity or Chernobyl or whatever is the current scare story in the newspapers. I suspect that it is not getting commoner at all - it is simply that we are recognising cases that we never used to find.
When I was young, everyday I looked at all the blood tests performed on everyone from a population of half a million. On a good day there were 300 blood counts. Thirty years later my responsibilities had diminished; I only looked at the blood counts from a quarter of a million. But most days there were over 700 tests. Thirty years ago you needed to have a lymphocyte count in the blood of 15,000 befor you could diagnose CLL. Today it is only 5000. Thirty years ago we had just discovered that CLL was a B cell neoplasm. Today we can distinguish several types of B cell neoplasm in the blood. We now know that 3.5% of people over 40 have a population of monoclonal B cells indistinguishable from CLL cells in their blood. The harder you look; the more you find.
The 25% in whom the CLL is not found by chance have a variety of symptoms and signs. (A symptom is something the patient feels; a sign is a physical abnormality the doctor finds.) The commonest finding is an enlarged lymph node.
Lymphocytes circulate through the body like other blood cells. For many their journey is just like other blood cells: out in the arteries, home in the veins. They have another circulation, though. The little thin-walled blood vessels that join the arteries to the veins are called capillaries. Lymphocytes can squeeze out through the walls of the capillaries and enter the tissues. From here they make their way home via the lymphatics, which are like veins for white blood cells. Lymph nodes are like stations on the lymphatic railway. They are normally small - about the size of a split pea - and organised. The job of lymphocytes is to fight against foreign invaders, and the lymph node is where they get their training and their orders before going into battle.
Lymph nodes enlarge during an immune response - you often feel 'glands' in your neck when you have a sore throat. In CLL they enlarge because the cells that are growing in an uncontrolled way normally live in the lymph nodes. In CLL we look for lymph nodes in particular places: the neck - we call these cervical nodes; the armpit - axillary nodes; and the groin - inguinal nodes. But there are lymph nodes all over the body and in CLL any lymph node can enlarge. Mostly they can't be felt, but there are some characteristic lymph nodes that are sometimes felt: submental nodes are under the point of the chin and epitrochlear nodes by the elbow. Lymph nodes at the back of the tummy - retroperitoneal nodes - cannot be felt with the hand but may be shown up by CT scan or ultrasound. Nodes in the chest also have to be visualized by some sort of imaging technique.
The spleen is about the size of a fist and is found deep under the ribs on the left of the tummy. It enlarges in CLL, but it has to get to three times its normal size before it can be felt. As it gets bigger it grows across the tummy towards the right groin. It seldom gets as big as this in CLL but it can grow this large in some kinds of hematological cancers. When it is examined by CT scan in CLL it is almost always found to be enlarged and patients think that they must therefore have advanced stage disease. This is misleading. As far as CLL is concerned a spleen is only considered to be enlarged if the doctor can feel it with his/her hand.
The spleen can be though of as a large lymph node attached to the blood circulation. But it also has other roles. It is the last resting place for dying red cells and platelets. When it enlarges it is apt to take out red cells and platelets before they are ready to go. This is known as hypersplenism and it results in anemia and thrombocytopenia.
Anemia means a reduction of the amount of hemoglobin in the blood (the abreviation in Hb not Hg; Hg is the abreviation for Mercury). Men and women have different normal ranges for Hb. For men it is 13.5 - 17 g/dL, for women 11.5 - 15 g/dL. Anemia cause tiredness, breathlessness, weakness, pale skin and mucous membranes and, in severe cases, the features of heart failure; but in many cases mild anemia has no signs or symptoms. Anemia in CLL is caused by suppression of the bone marrow by the infiltrating lymphocytes, or by hemolysis - the distruction of red cells by an antibody, or by hypersplenism. Of course CLL patients can become anemic for reasons unrelated to CLL, such as iron deficiency or pernicious anemia.
Thrombocytopenia means a reduction of the platelets in the blood. The normal range for platelets is 150,000 to 400,000 per cubic millimeter. The features of thrombocytopenia are bleeding and bruising. Platelets are there to plug holes in broken blood vessels and to help the blood to clot. Bleeding into the skin is called purpura and there are two types: petechiae are tiny pinhead sized blood spots; echymoses are anything larger. Thrombocytopenia can have the same causes as anemia: marrow infiltration, immune destruction and hypersplenism.
Patients may also suffer from so-called B symptoms. B symptoms come from Hodgkin's disease doctors who used them to decide whether to treat with chemotherapy or radiotherapy and they have been imported into CLL theology where they don't necessarily fit precisely. There are three types of B symptom: fever, weight loss and night sweats. Some doctors and most patients think that fatigue ahould also be a B symptom in CLL. I shall write about fatigue on another occasion.
Patients with CLL are more susceptible to infections that normal - they have an immunodeficiency. I will write about this in detail later.
To summarize: Most patients with CLL are discovered because they have a blood test for some other reason. If patients have signs or symptoms, the commonest is enlarged lymph nodes, but the spleen (or more rarely the liver) may also be enlarged. In the most severe cases there may be anemia or thrombocytopenia. Some patients have B symptoms and all have a degree of immunodeficiency.
Monday, December 12, 2005
What is chronic lymphocytic leukemia?
Almost every review begins "CLL is the commonest leukemia in the western world". I wonder who first wrote that sentence? And is it true?
Traditionally, it is believed that the great German pathologist Rudolf Virchow coined the term leukemia. It just means white blood. In fact the first description of leukemia (and I shall stick to the American spelling because it saves a key stroke) was by John Hughes Bennett, the Englishman who was Professor of Medicine at Edinburgh, a Scottish University, and who beat Virchow to the punch by 6 weeks. You can actually trace an even earlier case of what was probably leukemia from the writings of the Parisian doctor, Armand Velpeau, eighteen years earlier in 1827. Both Virchow and Bennett studied in Paris before they made their discoveries.
There are lots of causes of increased white cells in the blood that are not leukemia. What makes a leukemia is the fact all the cells are the children of a single parent. They are monoclonal - genetically identical members of the same clone. In CLL they are also lymphocytes. A lymphocyte is one of the 5 types of white blood cell (the others are monocyte, neutrophil, eosinophil and basophil). Lymphocytes are members of the immune system and are further subdivided into T cells (derived from the Thymus), B cells (derived from the Bone marrow) and NK cells (it stands for Natural Killer). CLL cells are B-lymphocytes.
(Immunologists will correct me here, because the B originally stood for the Bursa of Fabricius, an organ near the cloaca of a chicken which modifies fowl B cells. But mice and men don't have one, so B is for Bone Marrow was adopted instead.)
B lymphocytes are the ones that are destined to make antibody. Antibodies are chemicals made by the body to react specifically with something that is foreign to the body. (I am not going to explain antibodies in this posting - I will get to them at a later date.)
CLL is sometimes called a tumor of B cells, but that is confusing because tumor means swelling and not every case of CLL has a swelling. It's medical shorthand for a growth.
When I trained, doctors went in for euphemisms a lot. Instead of calling something a cancer it was called a growth or a neoplasm or a tumor or a mitotic lesion, or just a lesion. It is not simply a matter of avoiding telling the patient the truth; there is also the matter of precision. To most doctors cancer is the lay-term for carcinoma. Cancer is Latin for a crab - it is one of the signs of the Zodiac. Carcinoma is Greek for crab. The Patricians speak Greek; the Plebians talk Latin. A carcinoma is a malignant neoplasm of epithelial tissue. Blood is not an epithelial tissue, so we get uncomfortable when patients talk about a cancer of the blood.
Neoplasm just means newly formed or moulded or grown (Greek again). So in lay terms a neoplasm is a new growth. It isn't necessary cancer; it could be a benign (good) or a malignant (nasty) growth. A lesion refers to any kind of tissue damage (it comes from the Latin for hurt). A mitotic lesion is one that is characterised by mitoses. Mitosis is the mechanism by which cells duplicate their chromosomes and divide, so a mitotic lesion is a hurt in which cell division is prominent - in other words a cancer. Strictly speaking leukemias should be called sarcomas. A sarcoma is a malignant growth of a non-epithelial tissue. Epthelial tissue is what covers the outside of the body. But you have to remember that the lining of the gut is the outside of the body; outside but inside if you see what I mean.
We used to have the term lymphosarcoma. This was another term coined by Virchow. In the Bible we read of God bringing all the animals to Adam in the Garden of Eden for him to give them names. Virchow was very like Adam, very good at naming things. Lymphosarcoma became to be used for a particular form of neoplasm of lymphocytes and is now no longer used. W use the term lymphoma instead to refer to neoplasms of lymphocytes and strictly speaking CLL is a form of lymphoma, though by convention we call liquid tumors leukemias and solid tumors lymphomas. When CLL is mainly solid it is known as small lymphocytic lymphoma or SLL. Often patients talk of having CLL/SLL. That usually means that they have seen an oncologist who is more comfortable dealing with lymphomas than leukemias.
"Chronic" is also from the Greek and has to do with time. We talk about Acute leukemias which happen quickly and Chronic leukemias that happen slowly.
So CLL is the slow malignant growth of one of the white blood cells - the one known as a B-lymphocyte that should have been in the business of making antibodies.
Traditionally, it is believed that the great German pathologist Rudolf Virchow coined the term leukemia. It just means white blood. In fact the first description of leukemia (and I shall stick to the American spelling because it saves a key stroke) was by John Hughes Bennett, the Englishman who was Professor of Medicine at Edinburgh, a Scottish University, and who beat Virchow to the punch by 6 weeks. You can actually trace an even earlier case of what was probably leukemia from the writings of the Parisian doctor, Armand Velpeau, eighteen years earlier in 1827. Both Virchow and Bennett studied in Paris before they made their discoveries.
There are lots of causes of increased white cells in the blood that are not leukemia. What makes a leukemia is the fact all the cells are the children of a single parent. They are monoclonal - genetically identical members of the same clone. In CLL they are also lymphocytes. A lymphocyte is one of the 5 types of white blood cell (the others are monocyte, neutrophil, eosinophil and basophil). Lymphocytes are members of the immune system and are further subdivided into T cells (derived from the Thymus), B cells (derived from the Bone marrow) and NK cells (it stands for Natural Killer). CLL cells are B-lymphocytes.
(Immunologists will correct me here, because the B originally stood for the Bursa of Fabricius, an organ near the cloaca of a chicken which modifies fowl B cells. But mice and men don't have one, so B is for Bone Marrow was adopted instead.)
B lymphocytes are the ones that are destined to make antibody. Antibodies are chemicals made by the body to react specifically with something that is foreign to the body. (I am not going to explain antibodies in this posting - I will get to them at a later date.)
CLL is sometimes called a tumor of B cells, but that is confusing because tumor means swelling and not every case of CLL has a swelling. It's medical shorthand for a growth.
When I trained, doctors went in for euphemisms a lot. Instead of calling something a cancer it was called a growth or a neoplasm or a tumor or a mitotic lesion, or just a lesion. It is not simply a matter of avoiding telling the patient the truth; there is also the matter of precision. To most doctors cancer is the lay-term for carcinoma. Cancer is Latin for a crab - it is one of the signs of the Zodiac. Carcinoma is Greek for crab. The Patricians speak Greek; the Plebians talk Latin. A carcinoma is a malignant neoplasm of epithelial tissue. Blood is not an epithelial tissue, so we get uncomfortable when patients talk about a cancer of the blood.
Neoplasm just means newly formed or moulded or grown (Greek again). So in lay terms a neoplasm is a new growth. It isn't necessary cancer; it could be a benign (good) or a malignant (nasty) growth. A lesion refers to any kind of tissue damage (it comes from the Latin for hurt). A mitotic lesion is one that is characterised by mitoses. Mitosis is the mechanism by which cells duplicate their chromosomes and divide, so a mitotic lesion is a hurt in which cell division is prominent - in other words a cancer. Strictly speaking leukemias should be called sarcomas. A sarcoma is a malignant growth of a non-epithelial tissue. Epthelial tissue is what covers the outside of the body. But you have to remember that the lining of the gut is the outside of the body; outside but inside if you see what I mean.
We used to have the term lymphosarcoma. This was another term coined by Virchow. In the Bible we read of God bringing all the animals to Adam in the Garden of Eden for him to give them names. Virchow was very like Adam, very good at naming things. Lymphosarcoma became to be used for a particular form of neoplasm of lymphocytes and is now no longer used. W use the term lymphoma instead to refer to neoplasms of lymphocytes and strictly speaking CLL is a form of lymphoma, though by convention we call liquid tumors leukemias and solid tumors lymphomas. When CLL is mainly solid it is known as small lymphocytic lymphoma or SLL. Often patients talk of having CLL/SLL. That usually means that they have seen an oncologist who is more comfortable dealing with lymphomas than leukemias.
"Chronic" is also from the Greek and has to do with time. We talk about Acute leukemias which happen quickly and Chronic leukemias that happen slowly.
So CLL is the slow malignant growth of one of the white blood cells - the one known as a B-lymphocyte that should have been in the business of making antibodies.
What is it here for?
The main purpose of this blog is to be a source of information for patients with chronic lymphocytic leukemia (CLL). In writing about this disease I shall let slip some things about myself. Can't be helped - and since it can't be helped I shall do it deliberately.
First: about the title. It is, of course, a play on the title of Wordsworth's famous poem "Intimations of Immortality". An early line from this long poem states, "The things which I have seen I now can see no more". This is a motive for writing. These days unless I write down what I think, I forget what I was thinking about. Whether it is worth remembering is for others to judge.
"Mutations" refers to the most significant piece of work I have done on CLL: the discovery that the mutational status of the immunoglobulin genes is the most important prognostic factor in this disease. But mutations are also changes, and changes are things that I will reflect on in this blog. One of Wordsworth's sonnets is entitled "Mutability" but his changes are all at the behest of the second law of thermodynamics: "entropy increases" or the housewife's version "things wear out". Change for Wordsworth is always an inability to sustain "the unimaginable touch of time". Are all changes for the worse? This we will discover as we read on.
Mortality comes to us all as long as (as they used to say) the Lord tarries. But not to CLL cells. A signal characteristic of the CLL cell is its immortality. Or in scientific-speak, its failure of apoptosis.
I used to joke that having CLL was a guarantee of immortality. What I meant was that by an large it is a disease of the old, and if you happen to have the mutated version the disease didn't kill you. Many of my patients were in their nineties and some were over a hundred. So perhaps it should be "Mutations of Immortality"? I experimented with this but I liked the alliteration and I wanted eventually to write about the problems of dying.
The word "leukemia" leaks terror in the staunchest chest. But this leukemia kills by stealth not strength. It hangs around for years and sometimes for decades. There is time to consider one's own mortality. And we have time.
First: about the title. It is, of course, a play on the title of Wordsworth's famous poem "Intimations of Immortality". An early line from this long poem states, "The things which I have seen I now can see no more". This is a motive for writing. These days unless I write down what I think, I forget what I was thinking about. Whether it is worth remembering is for others to judge.
"Mutations" refers to the most significant piece of work I have done on CLL: the discovery that the mutational status of the immunoglobulin genes is the most important prognostic factor in this disease. But mutations are also changes, and changes are things that I will reflect on in this blog. One of Wordsworth's sonnets is entitled "Mutability" but his changes are all at the behest of the second law of thermodynamics: "entropy increases" or the housewife's version "things wear out". Change for Wordsworth is always an inability to sustain "the unimaginable touch of time". Are all changes for the worse? This we will discover as we read on.
Mortality comes to us all as long as (as they used to say) the Lord tarries. But not to CLL cells. A signal characteristic of the CLL cell is its immortality. Or in scientific-speak, its failure of apoptosis.
I used to joke that having CLL was a guarantee of immortality. What I meant was that by an large it is a disease of the old, and if you happen to have the mutated version the disease didn't kill you. Many of my patients were in their nineties and some were over a hundred. So perhaps it should be "Mutations of Immortality"? I experimented with this but I liked the alliteration and I wanted eventually to write about the problems of dying.
The word "leukemia" leaks terror in the staunchest chest. But this leukemia kills by stealth not strength. It hangs around for years and sometimes for decades. There is time to consider one's own mortality. And we have time.
Subscribe to:
Comments (Atom)